Department of Computer Science and Information Engineering, National Cheng Kung University, No. 1, University Road, Tainan City 701, Taiwan R.O.C.
BMC Bioinformatics. 2013 Jul 21;14:230. doi: 10.1186/1471-2105-14-230.
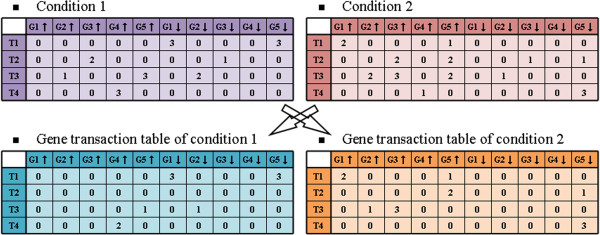
Frequent pattern mining analysis applied on microarray dataset appears to be a promising strategy for identifying relationships between gene expression levels. Unfortunately, too many itemsets (co-expressed genes) are identified by this analysis method since it does not consider the importance of each gene within biological processes to a cellular response and does not take into account temporal properties under biological treatment-control matched conditions in a microarray dataset.
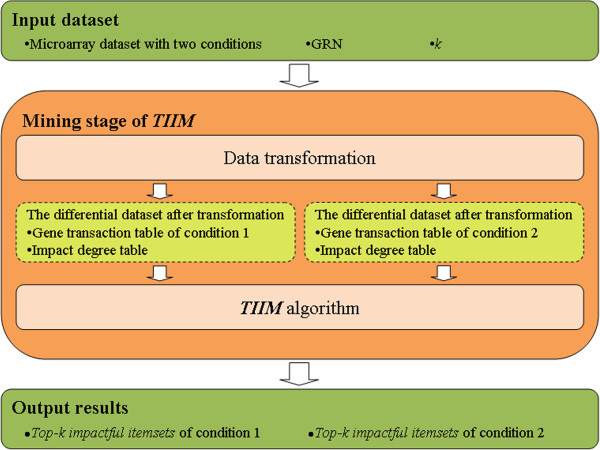
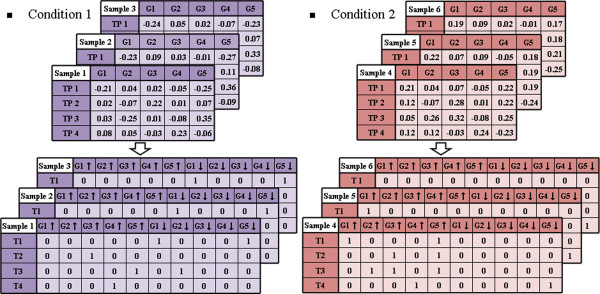
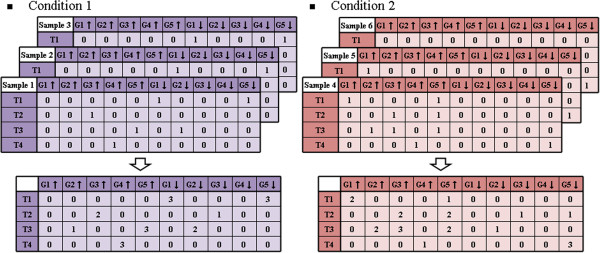
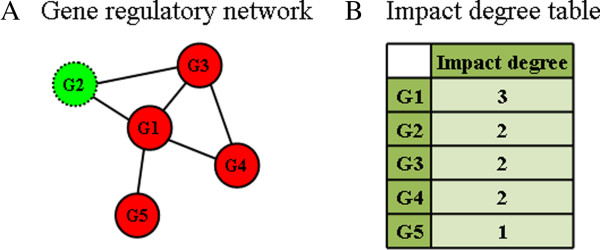
We propose a method termed TIIM (Top-k Impactful Itemsets Miner), which only requires specifying a user-defined number k to explore the top k itemsets with the most significantly differentially co-expressed genes between 2 conditions in a time course. To give genes different weights, a table with impact degrees for each gene was constructed based on the number of neighboring genes that are differently expressed in the dataset within gene regulatory networks. Finally, the resulting top-k impactful itemsets were manually evaluated using previous literature and analyzed by a Gene Ontology enrichment method.
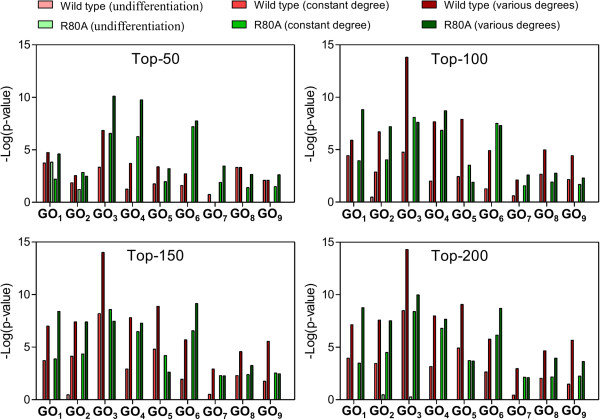
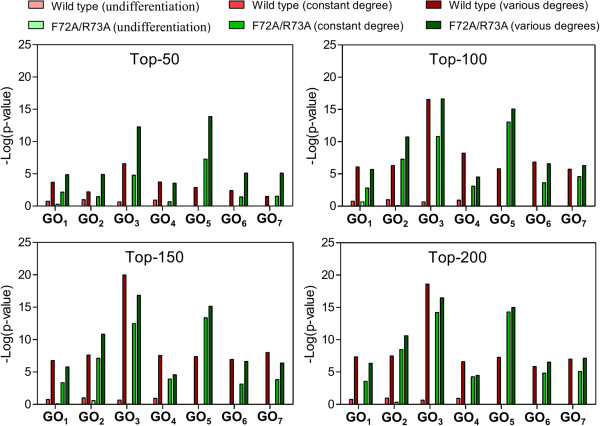
In this study, the proposed method was evaluated in 2 publicly available time course microarray datasets with 2 different experimental conditions. Both datasets identified potential itemsets with co-expressed genes evaluated from the literature and showed higher accuracies compared to the 2 corresponding control methods: i) performing TIIM without considering the gene expression differentiation between 2 different experimental conditions and impact degrees, and ii) performing TIIM with a constant impact degree for each gene. Our proposed method found that several new gene regulations involved in these itemsets were useful for biologists and provided further insights into the mechanisms underpinning biological processes. The Java source code and other related materials used in this study are available at "http://websystem.csie.ncku.edu.tw/TIIM_Program.rar".
在微阵列数据集上应用频繁模式挖掘分析似乎是识别基因表达水平之间关系的一种很有前途的策略。不幸的是,由于该分析方法不考虑基因在细胞反应中对生物过程的重要性,也不考虑微阵列数据集中生物处理对照匹配条件下的时间特性,因此会识别出太多的项集(共表达基因)。
我们提出了一种称为 TIIM(Top-k Impactful Itemsets Miner)的方法,该方法仅需要指定用户定义的数量 k,即可在时间序列中探索 2 种条件之间具有最显著差异共表达基因的前 k 个项集。为了给基因赋予不同的权重,根据基因调控网络中数据集内差异表达的相邻基因的数量,构建了一个包含每个基因影响程度的表。最后,使用先前的文献手动评估生成的 top-k 影响项集,并通过基因本体论富集方法进行分析。
在这项研究中,使用 2 个具有 2 种不同实验条件的公开可用的时间序列微阵列数据集对所提出的方法进行了评估。这两个数据集都从文献中评估了具有共表达基因的潜在项集,并且与 2 个相应的对照方法相比,准确性更高:i)不考虑 2 种不同实验条件和影响程度的情况下执行 TIIM,ii)为每个基因执行 TIIM 时使用恒定的影响程度。我们提出的方法发现,这些项集中涉及的几个新的基因调控对生物学家很有用,并为理解生物过程背后的机制提供了更深入的见解。本研究中使用的 Java 源代码和其他相关材料可在“http://websystem.csie.ncku.edu.tw/TIIM_Program.rar”处获得。