Université Pierre et Marie Curie (UPMC), UMR 7139 Végétaux marins et Biomolécules, Station Biologique, CS 90074, F29688, Roscoff, France and Centre National de la Recherche Scientifique (CNRS), UMR 7139 Végétaux marins et Biomolécules, Station Biologique, CS 90074, F29688, Roscoff, France.
Nucleic Acids Res. 2014 Jan;42(1):417-29. doi: 10.1093/nar/gkt856. Epub 2013 Sep 26.
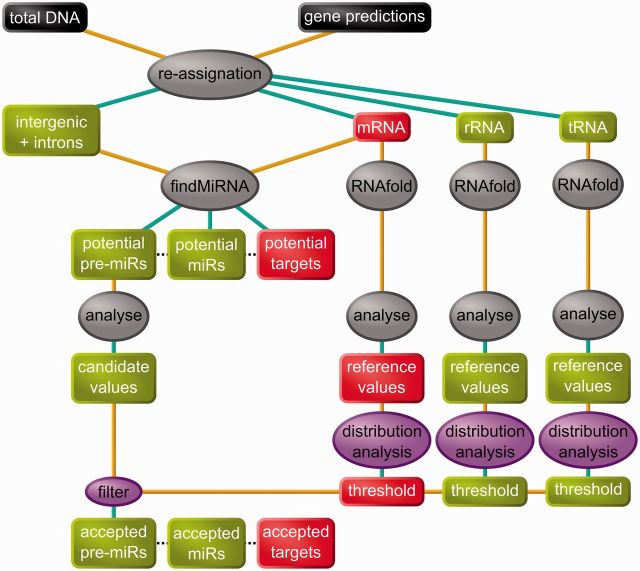
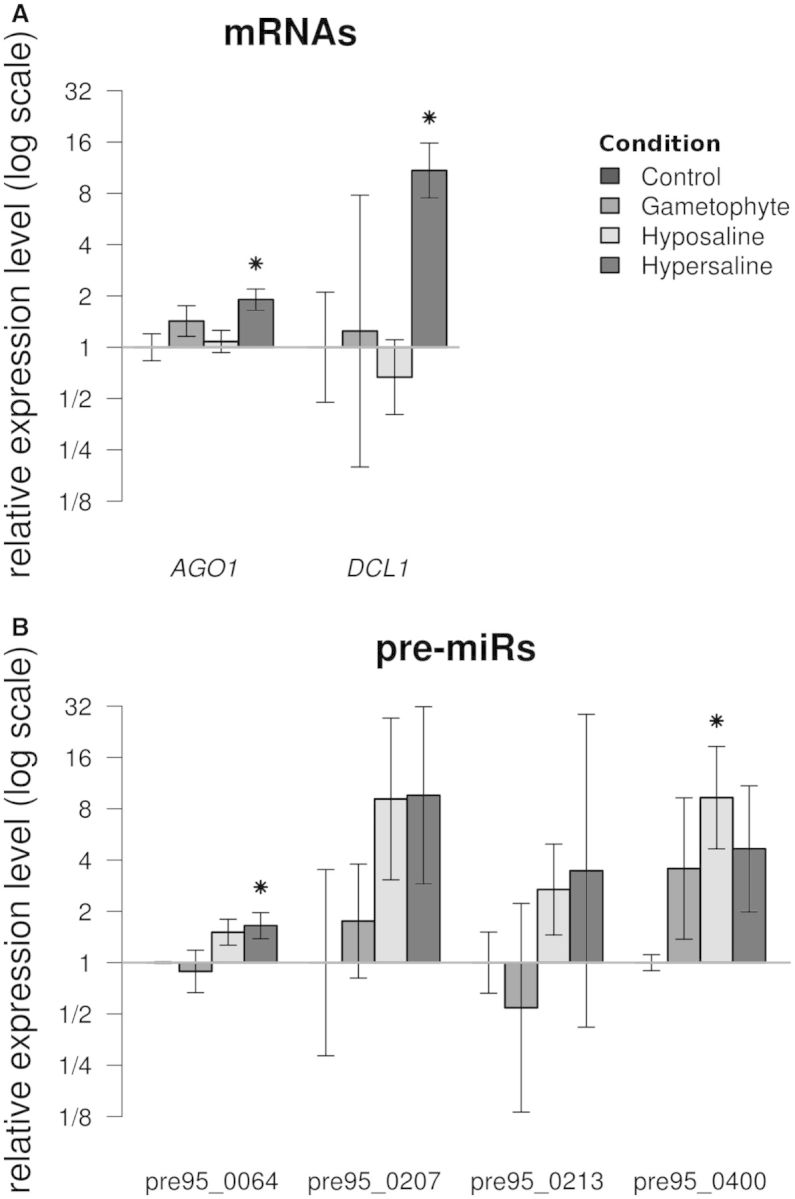
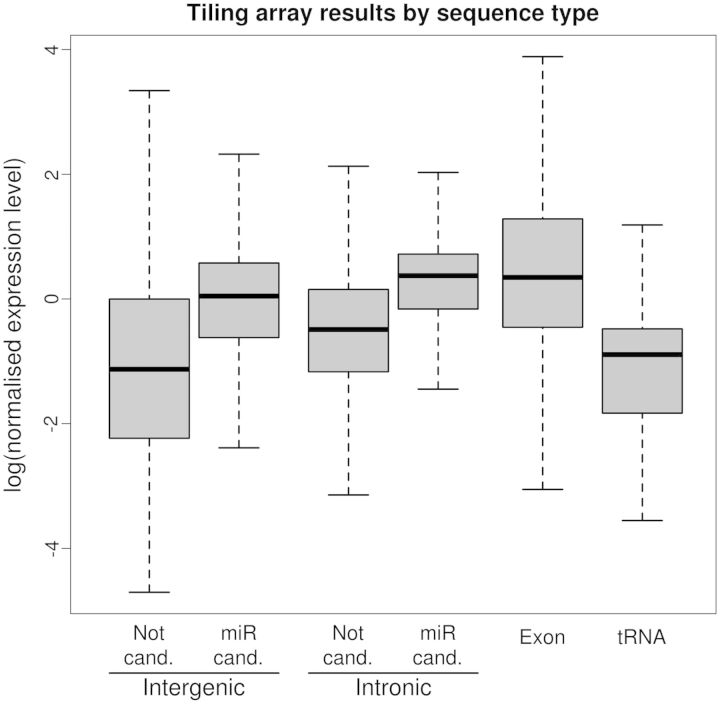
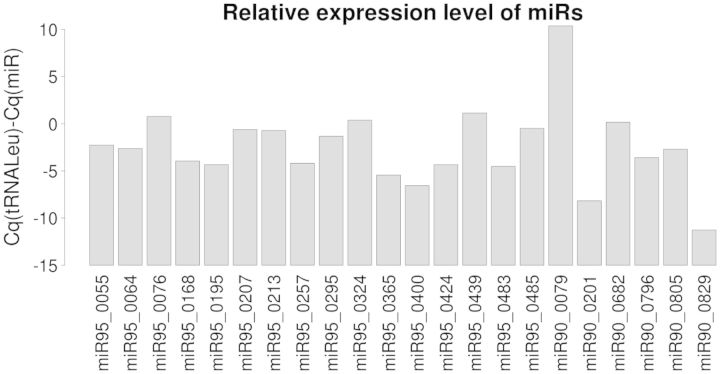
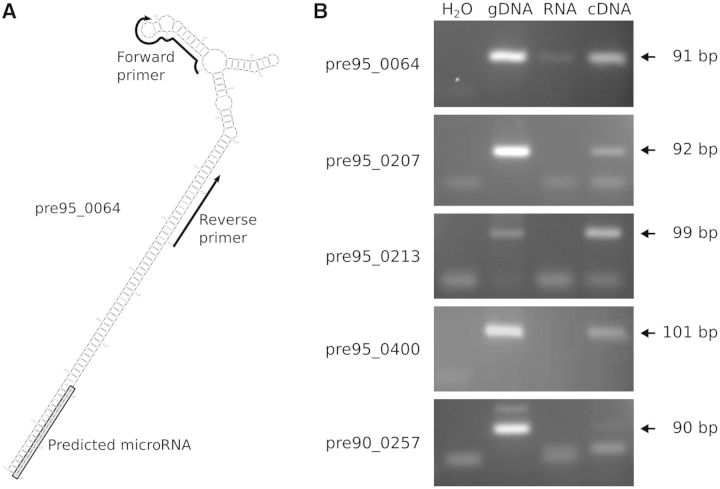
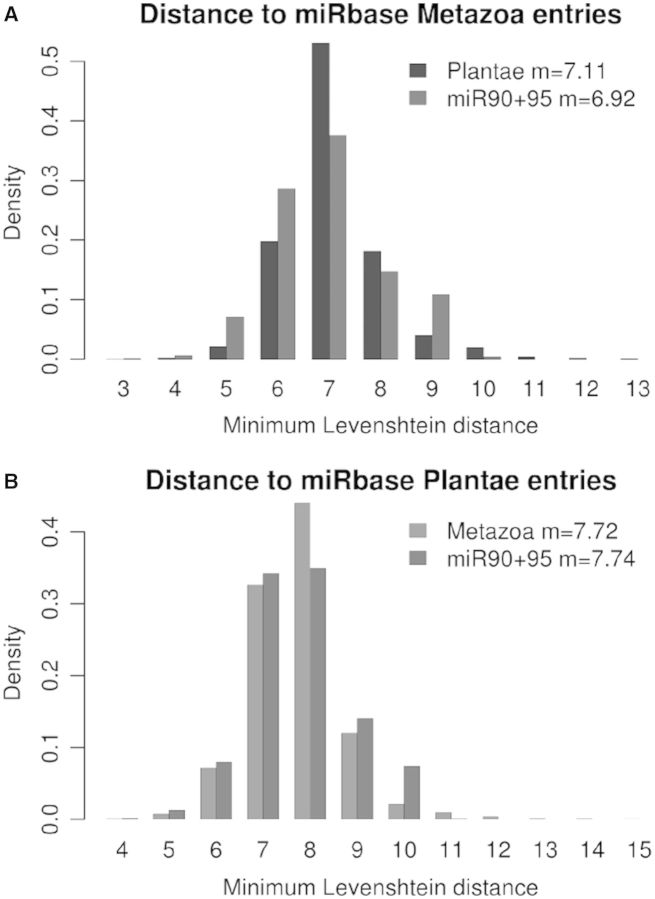
We used an in silico approach to predict microRNAs (miRNAs) genome-wide in the brown alga Ectocarpus siliculosus. As brown algae are phylogenetically distant from both animals and land plants, our approach relied on features shared by all known organisms, excluding sequence conservation, genome localization and pattern of base-pairing with the target. We predicted between 500 and 1500 miRNAs candidates, depending on the values of the energetic parameters used to filter the potential precursors. Using quantitative polymerase chain reaction assays, we confirmed the existence of 22 miRNAs among 72 candidates tested, and of 8 predicted precursors. In addition, we compared the expression of miRNAs and their precursors in two life cycle states (sporophyte, gametophyte) and under salt stress. Several miRNA precursors, Argonaute and DICER messenger RNAs were differentially expressed in these conditions. Finally, we analyzed the gene organization and the target functions of the predicted candidates. This showed that E. siliculosus miRNA genes are, like plant miRNA genes, rarely clustered and, like animal miRNA genes, often located in introns. Among the predicted targets, several widely conserved functional domains are significantly overrepresented, like kinesin, nucleotide-binding/APAF-1, R proteins and CED-4 (NB-ARC) and tetratricopeptide repeats. The combination of computational and experimental approaches thus emphasizes the originality of molecular and cellular processes in brown algae.
我们采用计算机模拟的方法,对褐藻(Ectocarpus siliculosus)中的 microRNAs (miRNAs) 进行了全基因组预测。由于褐藻在系统发生上与动物和陆地植物都相距甚远,因此我们的方法依赖于所有已知生物共有的特征,不包括序列保守性、基因组定位和与靶标碱基配对的模式。我们根据用于过滤潜在前体的能量参数值,预测了 500 到 1500 个 miRNA 候选物。通过定量聚合酶链反应 (qPCR) 检测,我们在 72 个候选 miRNA 中证实了 22 个 miRNA 的存在,并预测了 8 个 miRNA 前体。此外,我们比较了在两种生命周期状态(孢子体、配子体)和盐胁迫下的 miRNA 和其前体的表达。在这些条件下,几个 miRNA 前体、Argonaute 和 DICER 信使 RNA 的表达水平不同。最后,我们分析了预测候选物的基因组织和靶基因功能。结果表明,E. siliculosus 的 miRNA 基因与植物 miRNA 基因一样,很少发生聚类,与动物 miRNA 基因一样,经常位于内含子中。在预测的靶基因中,几个广泛保守的功能结构域显著过表达,如驱动蛋白、核苷酸结合/APAF-1、R 蛋白和 CED-4 (NB-ARC) 以及 tetratricopeptide 重复。因此,计算和实验方法的结合强调了褐藻中分子和细胞过程的独特性。