ASTAR Institute of Molecular & Cell Biology, Proteos, Singapore ; Department of Biological Sciences, National University of Singapore, Singapore.
PLoS Genet. 2013;9(12):e1003955. doi: 10.1371/journal.pgen.1003955. Epub 2013 Dec 5.
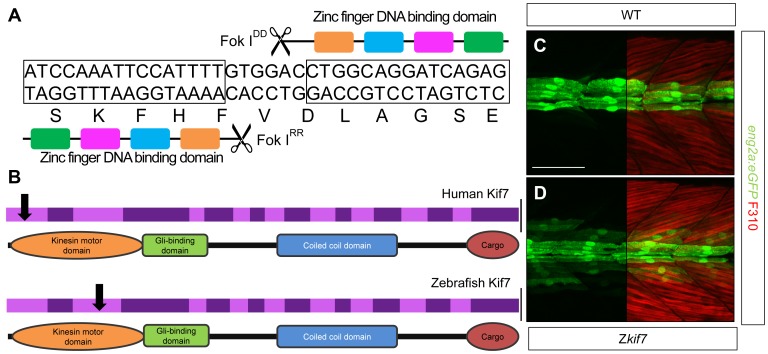
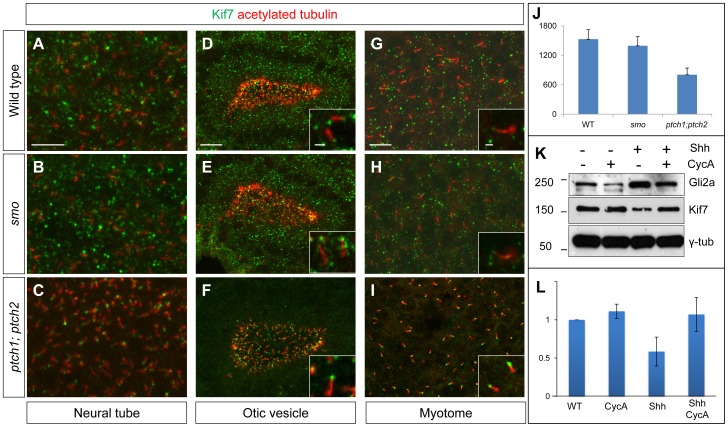
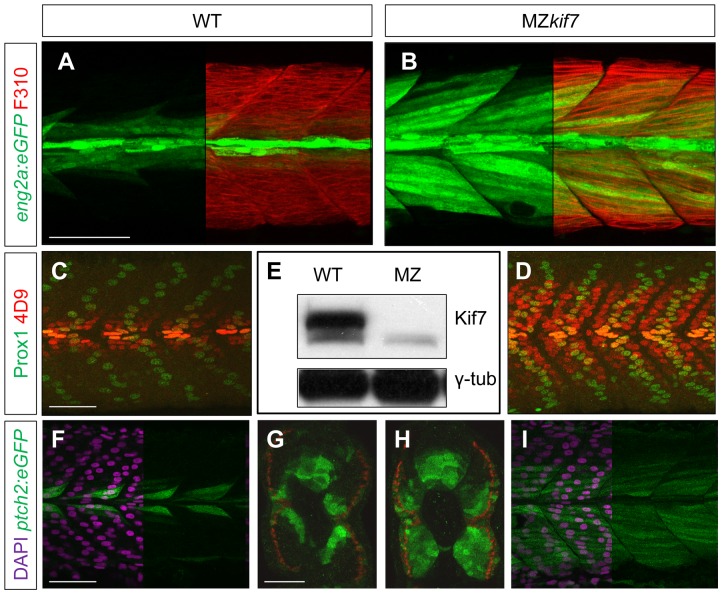
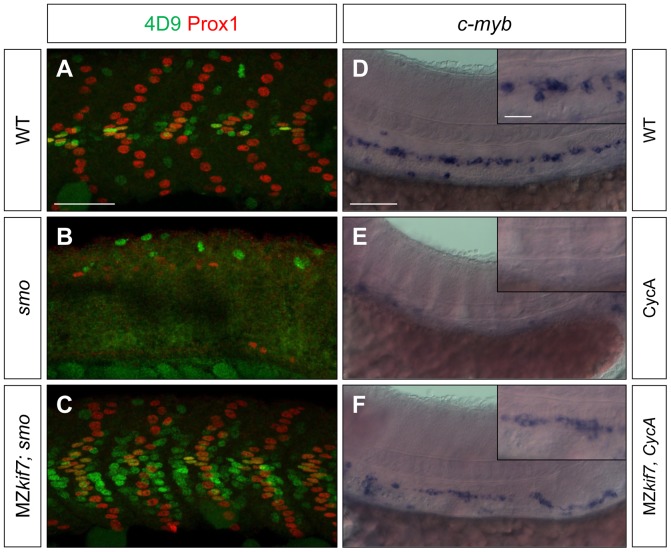
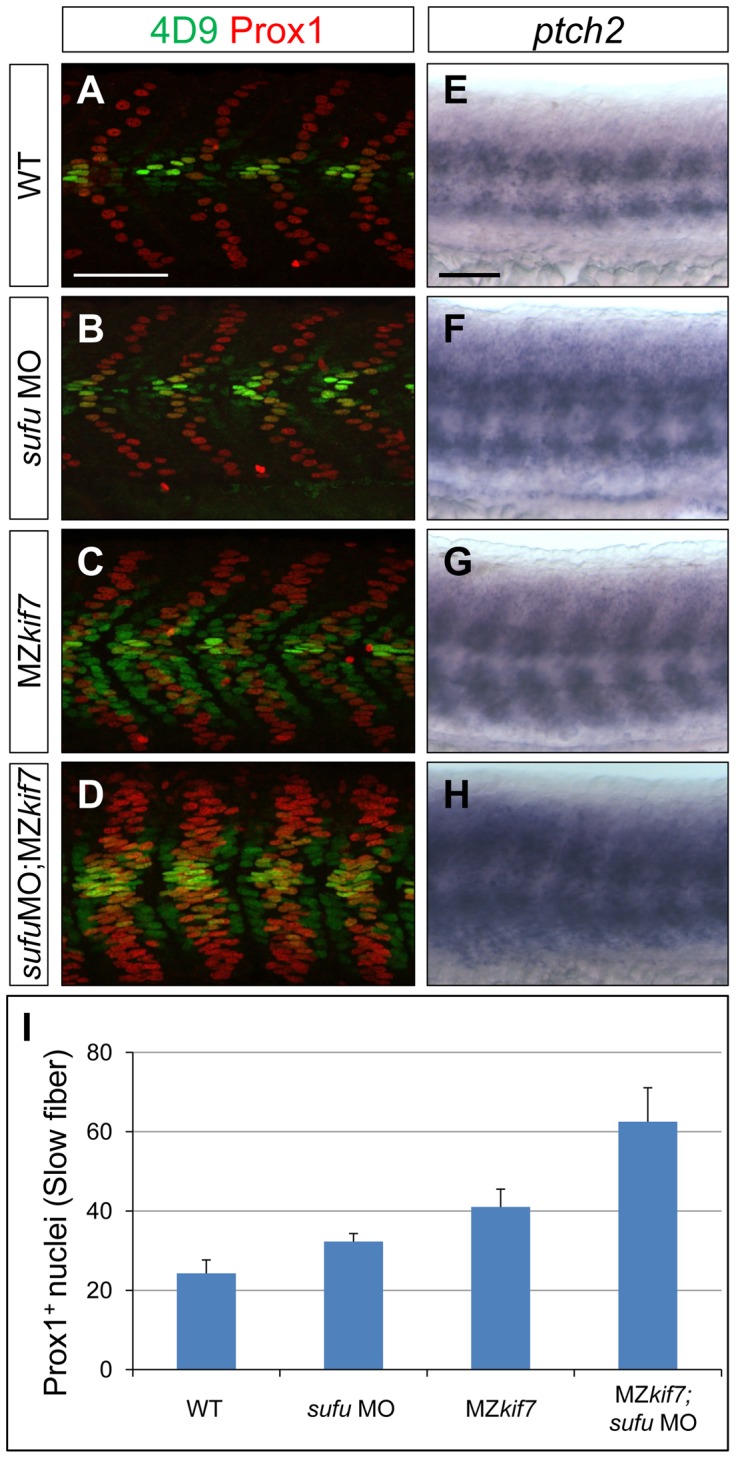
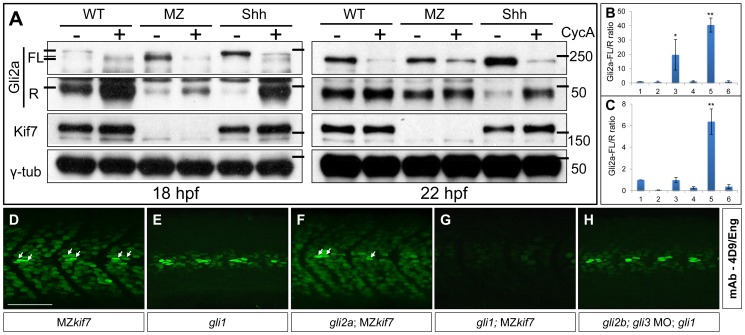
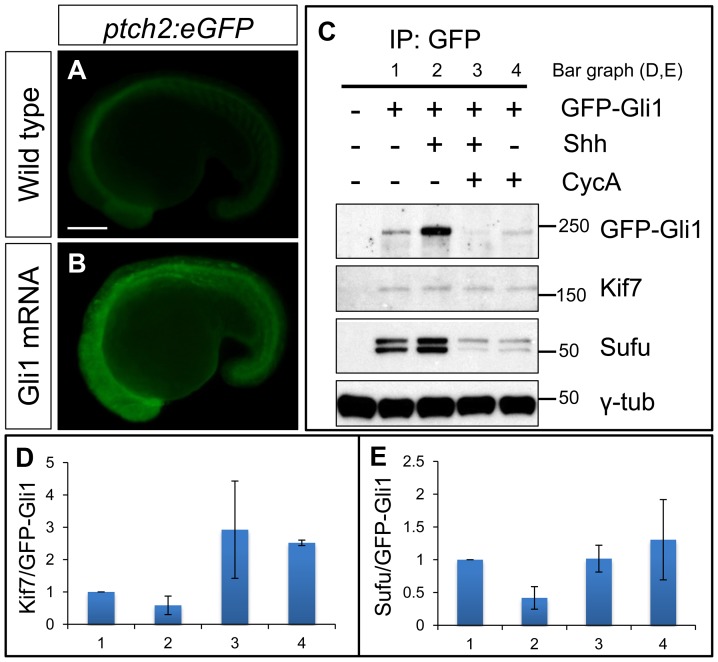
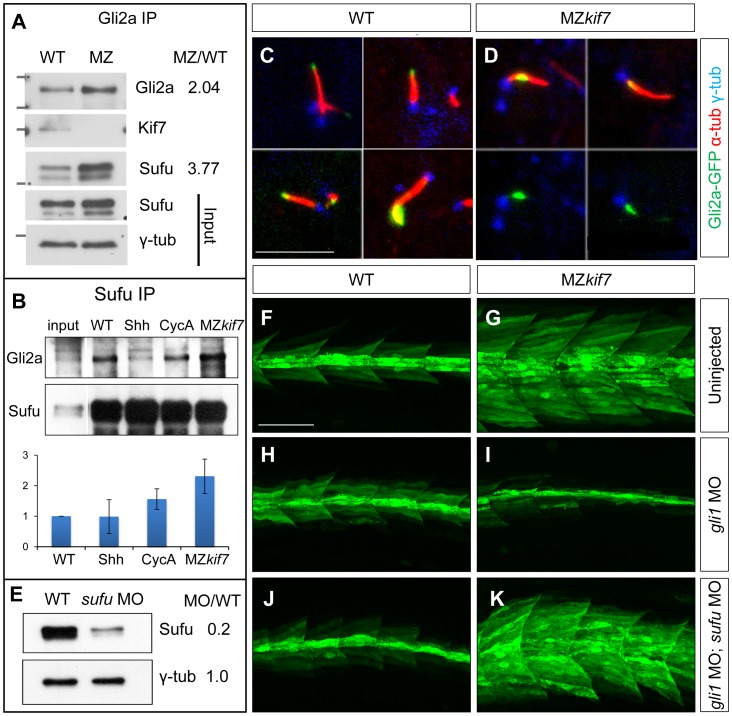
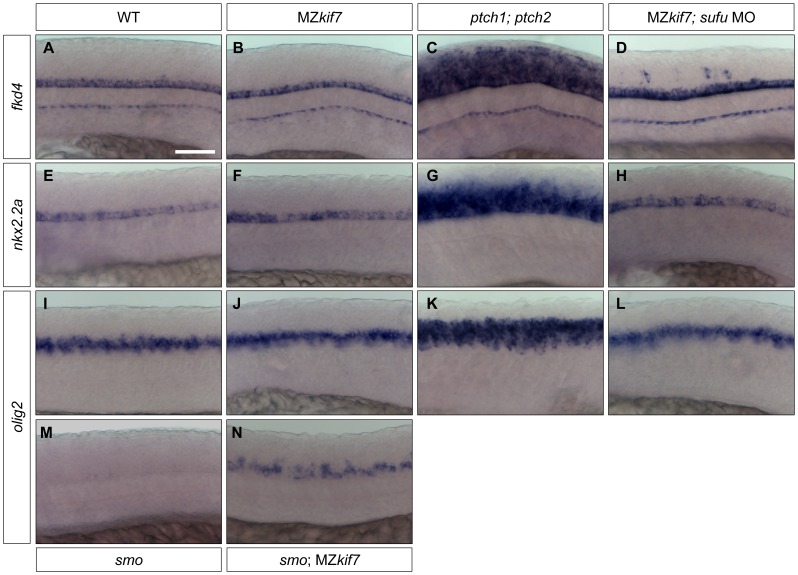
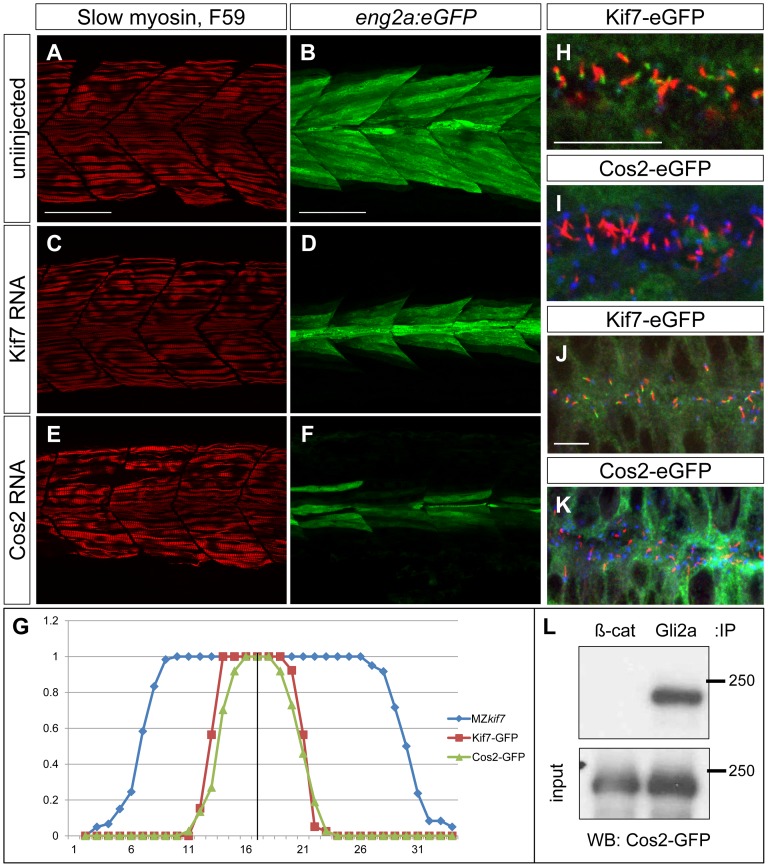
Loss of function mutations of Kif7, the vertebrate orthologue of the Drosophila Hh pathway component Costal2, cause defects in the limbs and neural tubes of mice, attributable to ectopic expression of Hh target genes. While this implies a functional conservation of Cos2 and Kif7 between flies and vertebrates, the association of Kif7 with the primary cilium, an organelle absent from most Drosophila cells, suggests their mechanisms of action may have diverged. Here, using mutant alleles induced by Zinc Finger Nuclease-mediated targeted mutagenesis, we show that in zebrafish, Kif7 acts principally to suppress the activity of the Gli1 transcription factor. Notably, we find that endogenous Kif7 protein accumulates not only in the primary cilium, as previously observed in mammalian cells, but also in cytoplasmic puncta that disperse in response to Hh pathway activation. Moreover, we show that Drosophila Costal2 can substitute for Kif7, suggesting a conserved mode of action of the two proteins. We show that Kif7 interacts with both Gli1 and Gli2a and suggest that it functions to sequester Gli proteins in the cytoplasm, in a manner analogous to the regulation of Ci by Cos2 in Drosophila. We also show that zebrafish Kif7 potentiates Gli2a activity by promoting its dissociation from the Suppressor of Fused (Sufu) protein and present evidence that it mediates a Smo dependent modification of the full length form of Gli2a. Surprisingly, the function of Kif7 in the zebrafish embryo appears restricted principally to mesodermal derivatives, its inactivation having little effect on neural tube patterning, even when Sufu protein levels are depleted. Remarkably, zebrafish lacking all Kif7 function are viable, in contrast to the peri-natal lethality of mouse kif7 mutants but similar to some Acrocallosal or Joubert syndrome patients who are homozygous for loss of function KIF7 alleles.
Kif7 的功能丧失突变,即果蝇 Hh 通路成分 Costal2 的脊椎动物同源物,导致小鼠的肢体和神经管缺陷,这归因于 Hh 靶基因的异位表达。虽然这表明 Cos2 和 Kif7 在苍蝇和脊椎动物之间具有功能上的保守性,但 Kif7 与初级纤毛的关联,即大多数果蝇细胞中不存在的细胞器,表明它们的作用机制可能已经分化。在这里,我们使用锌指核酸酶介导的靶向诱变诱导的突变等位基因,表明在斑马鱼中,Kif7 主要作用是抑制 Gli1 转录因子的活性。值得注意的是,我们发现内源性 Kif7 蛋白不仅在初级纤毛中积累,如以前在哺乳动物细胞中观察到的,而且在细胞质点状结构中积累,这些点状结构在 Hh 通路激活时分散。此外,我们表明果蝇 Costal2 可以替代 Kif7,表明这两种蛋白质具有保守的作用模式。我们表明 Kif7 与 Gli1 和 Gli2a 相互作用,并表明它通过将 Gli 蛋白在细胞质中隔离来发挥作用,类似于 Costal2 在果蝇中对 Ci 的调控。我们还表明,斑马鱼 Kif7 通过促进其从融合抑制因子 (Sufu) 蛋白解离来增强 Gli2a 的活性,并提供证据表明它介导Gli2a 全长形式的 Smo 依赖性修饰。令人惊讶的是,Kif7 在斑马鱼胚胎中的功能似乎主要局限于中胚层衍生物,其失活对神经管模式形成几乎没有影响,即使 Sufu 蛋白水平耗尽。值得注意的是,与小鼠 kif7 突变体的围产期致死性相反,缺乏所有 Kif7 功能的斑马鱼是存活的,但与一些具有功能丧失 KIF7 等位基因的常染色体显性或 Joubert 综合征患者相似。