Department of Pathology and Pathophysiology, Medical College of Soochow University, Suzhou, Jiangsu, China.
Department of Forensic Medicine, Medical College of Soochow University, Suzhou, Jiangsu, China.
PLoS One. 2013 Dec 23;8(12):e83072. doi: 10.1371/journal.pone.0083072. eCollection 2013.
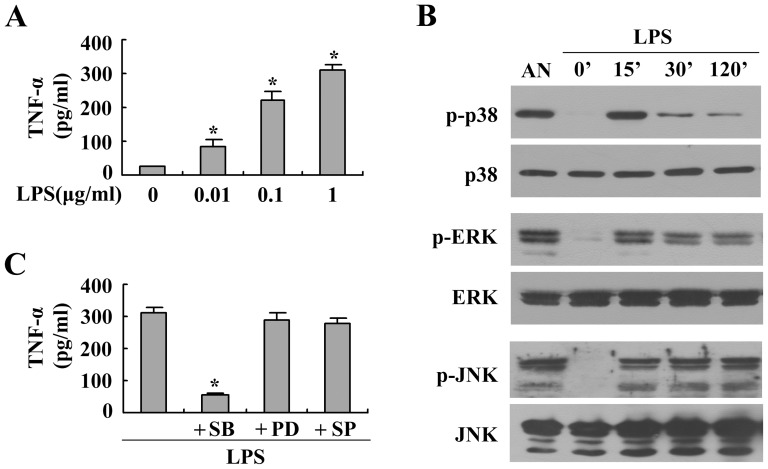
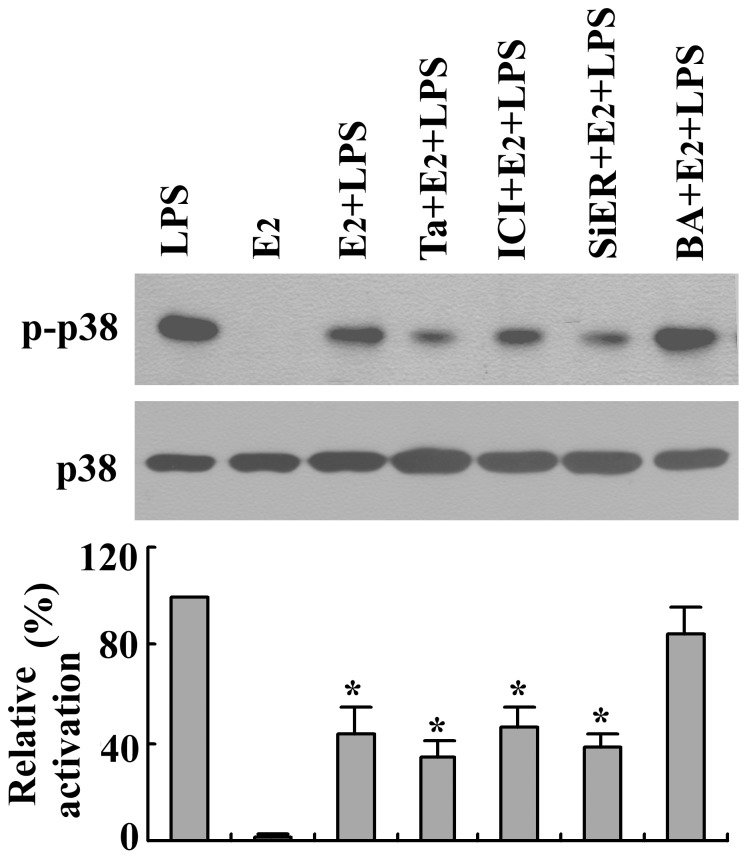
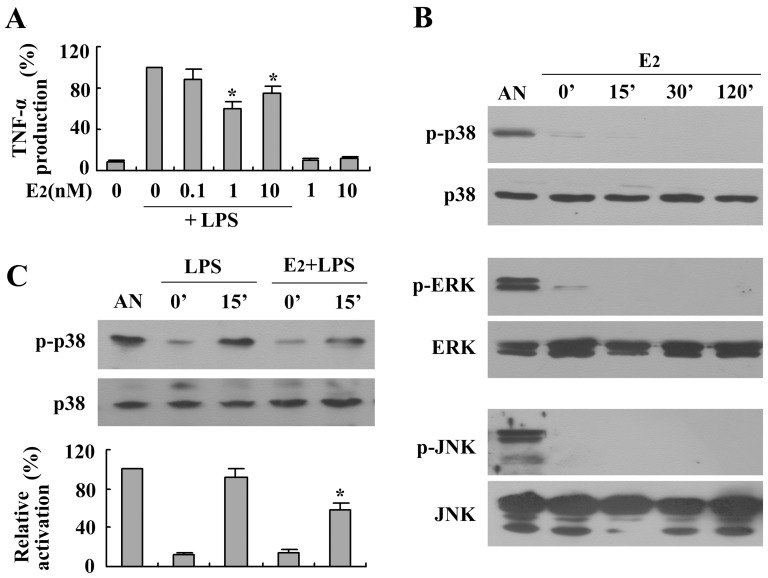
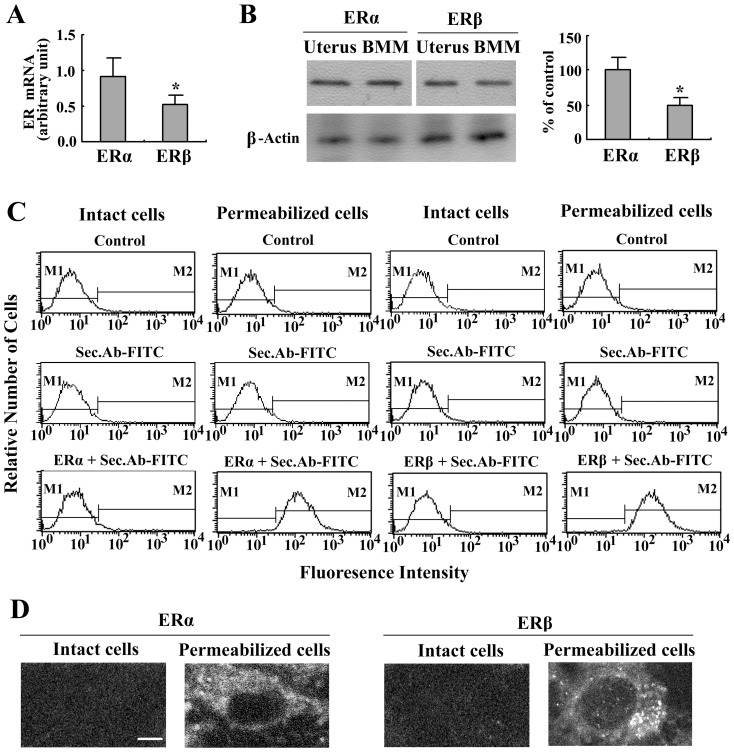
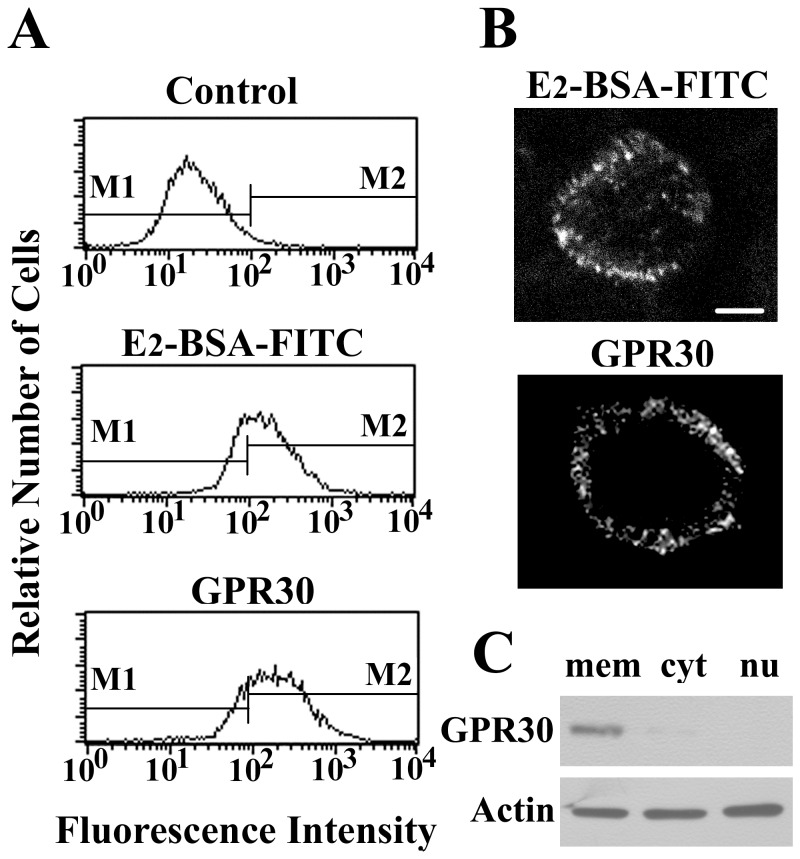
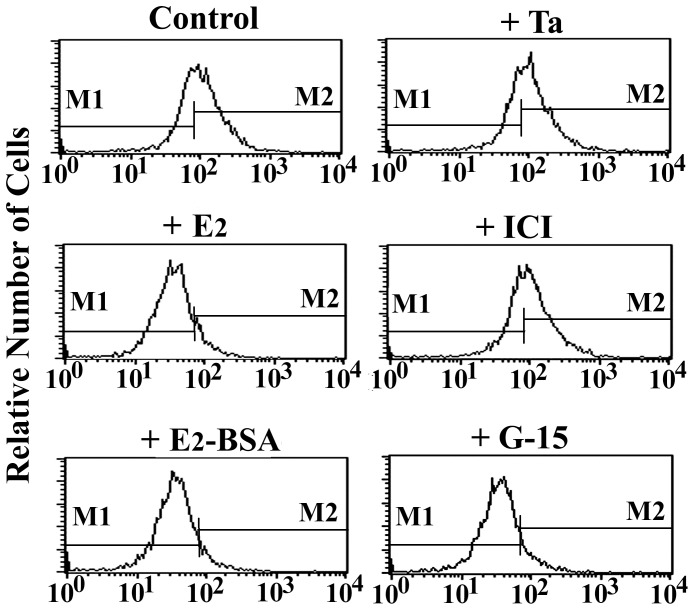
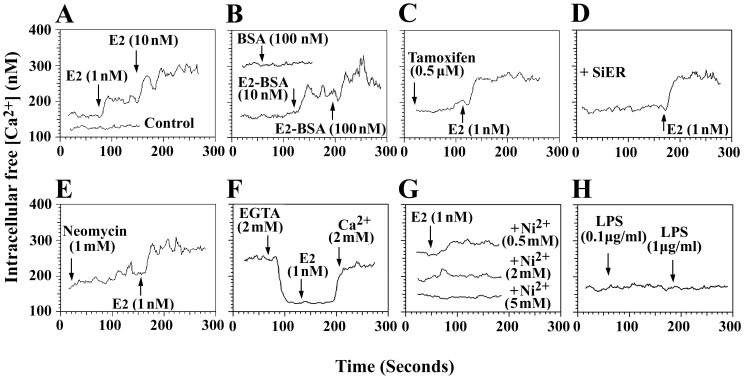
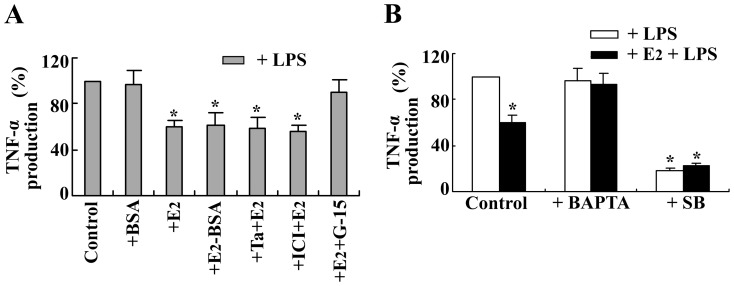
Estrogen is traditionally thought to exert genomic actions through members of the nuclear receptor family. Here, we investigated the rapid nongenomic effects of 17β-estradiol (E2) on tumor necrosis factor α (TNF-α) production following lipopolysaccharide (LPS) stimulation in mouse bone marrow-derived macrophages (BMMs). We found that LPS induced TNF-α production in BMMs via phosphorylation of p38 mitogen-activated protein kinase (MAPK). E2 itself did not affect the MAPK pathway, although it attenuated LPS-induced TNF-α production through suppression of p38 MAPK activation. Recently, G protein-coupled receptor 30 (GPR30) was suggested to be a membrane estrogen receptor (mER) that can mediate nongenomic estradiol signaling. We found that BMMs expressed both intracellular estrogen receptors (iER) and mER GPR30. The specific GPR30 antagonist G-15 significantly blocked effects of estradiol on LPS-induced TNF-α production, whereas an iER antagonist did not. Moreover, E2 induced a rapid rise in intracellular free Ca(2+) that was due to the influx of extracellular Ca(2+) and was not inhibited by an iER antagonist or silencing of iER. Ca(2+) influx was also induced by an impermeable E2 conjugated to BSA (E2-BSA), which has been used to investigate the nongenomic effects of estrogen. Consequently, Ca(2+), a pivotal factor in E2-stimulated nongenomic action, was identified as the key mediator. The inhibitory effects of E2 on LPS-induced TNF-α production and p38 MAPK phosphorylation were dependent on E2-triggered Ca(2+) influx because BAPTA, an intracellular Ca(2+) chelator, prevented these effects. Taken together, these data indicate that E2 can down-regulate LPS-induced TNF-α production via blockade of p38 MAPK phosphorylation through the mER-mediated nongenomic Ca(2+) signaling pathway in BMMs.
雌激素传统上被认为通过核受体家族成员发挥基因组作用。在这里,我们研究了 17β-雌二醇(E2)在脂多糖(LPS)刺激后对小鼠骨髓来源的巨噬细胞(BMM)中肿瘤坏死因子α(TNF-α)产生的快速非基因组效应。我们发现 LPS 通过磷酸化丝裂原活化蛋白激酶(MAPK)p38 诱导 BMM 中 TNF-α 的产生。E2 本身不会影响 MAPK 途径,尽管它通过抑制 p38 MAPK 激活来减弱 LPS 诱导的 TNF-α 产生。最近,G 蛋白偶联受体 30(GPR30)被认为是一种膜雌激素受体(mER),可以介导非基因组雌二醇信号。我们发现 BMM 表达细胞内雌激素受体(iER)和 mER GPR30。特异性 GPR30 拮抗剂 G-15 显著阻断了雌二醇对 LPS 诱导的 TNF-α 产生的作用,而 iER 拮抗剂则没有。此外,E2 诱导细胞内游离 Ca(2+)的快速增加,这是由于细胞外 Ca(2+)的流入引起的,并且不受 iER 拮抗剂或 iER 沉默的抑制。E2 与 BSA 结合的不可渗透 E2(E2-BSA)也诱导了 Ca(2+)流入,E2-BSA 已被用于研究雌激素的非基因组效应。因此,Ca(2+)被确定为 E2 刺激非基因组作用的关键介质。E2 对 LPS 诱导的 TNF-α 产生和 p38 MAPK 磷酸化的抑制作用依赖于 E2 触发的 Ca(2+)流入,因为细胞内 Ca(2+)螯合剂 BAPTA 可阻止这些作用。总之,这些数据表明,E2 可以通过 mER 介导的非基因组 Ca(2+)信号通路阻断 p38 MAPK 磷酸化来下调 LPS 诱导的 TNF-α 产生。