Department of Neurological Surgery, Room 502C, UC Davis, 1515 Newton Court, Davis, CA 95618, USA.
Department Clinical Genetics, Erasmus MC, Rotterdam, Netherlands.
J Neurodev Disord. 2014;6(1):25. doi: 10.1186/1866-1955-6-25. Epub 2014 Jul 30.
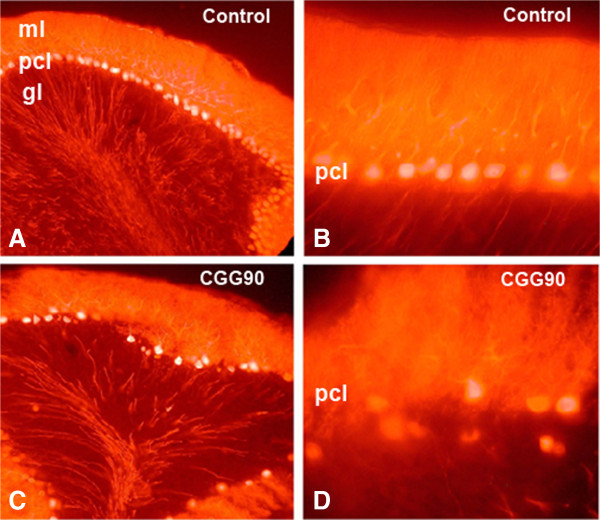
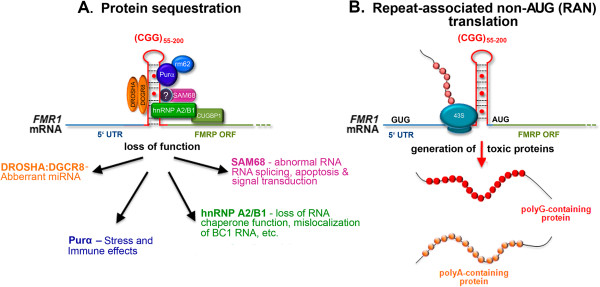
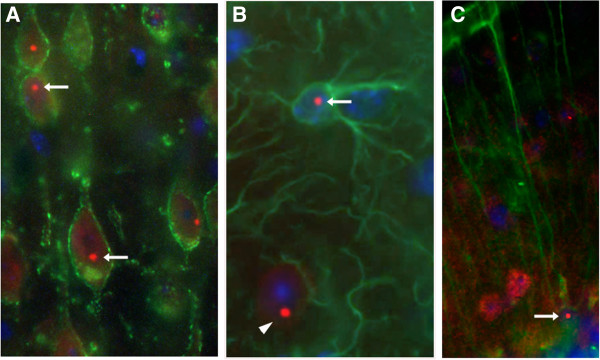
Carriers of the fragile X premutation (FPM) have CGG trinucleotide repeat expansions of between 55 and 200 in the 5'-UTR of FMR1, compared to a CGG repeat length of between 5 and 54 for the general population. Carriers were once thought to be without symptoms, but it is now recognized that they can develop a variety of early neurological symptoms as well as being at risk for developing the late onset neurodegenerative disorder fragile X-associated tremor/ataxia syndrome (FXTAS). Several mouse models have contributed to our understanding of FPM and FXTAS, and findings from studies using these models are summarized here. This review also discusses how this information is improving our understanding of the molecular and cellular abnormalities that contribute to neurobehavioral features seen in some FPM carriers and in patients with FXTAS. Mouse models show much of the pathology seen in FPM carriers and in individuals with FXTAS, including the presence of elevated levels of Fmr1 mRNA, decreased levels of fragile X mental retardation protein, and ubiquitin-positive intranuclear inclusions. Abnormalities in dendritic spine morphology in several brain regions are associated with neurocognitive deficits in spatial and temporal memory processes, impaired motor performance, and altered anxiety. In vitro studies have identified altered dendritic and synaptic architecture associated with abnormal Ca(2+) dynamics and electrical network activity. FPM mice have been particularly useful in understanding the roles of Fmr1 mRNA, fragile X mental retardation protein, and translation of a potentially toxic polyglycine peptide in pathology. Finally, the potential for using these and emerging mouse models for preclinical development of therapies to improve neurological function in FXTAS is considered.
脆性 X 前突变 (FPM) 携带者的 FMR1 5'UTR 中 CGG 三核苷酸重复扩展介于 55 至 200 之间,而普通人群的 CGG 重复长度介于 5 至 54 之间。曾经认为携带者没有症状,但现在人们认识到,他们可能会出现各种早期神经症状,并面临发生晚发性神经退行性疾病脆性 X 相关震颤共济失调综合征 (FXTAS) 的风险。几种小鼠模型有助于我们了解 FPM 和 FXTAS,这里总结了使用这些模型进行的研究结果。本文还讨论了这些信息如何提高我们对导致一些 FPM 携带者和 FXTAS 患者出现神经行为特征的分子和细胞异常的理解。小鼠模型显示出与 FPM 携带者和 FXTAS 个体中所见的许多病理学相似之处,包括 Fmr1 mRNA 水平升高、脆性 X 智力低下蛋白水平降低以及泛素阳性核内包涵体的存在。几个脑区树突棘形态的异常与空间和时间记忆过程中的神经认知缺陷、运动表现受损以及焦虑改变有关。体外研究已经确定了与异常 Ca(2+)动力学和电网络活动相关的树突和突触结构的改变。FPM 小鼠在理解 Fmr1 mRNA、脆性 X 智力低下蛋白以及潜在毒性多聚甘氨酸肽翻译在病理学中的作用方面特别有用。最后,考虑了使用这些和新兴的小鼠模型进行 FXTAS 神经功能改善的临床前治疗开发的潜力。