Ghosh Sharmila, Qu Zhipeng, Das Pranab J, Fang Erica, Juras Rytis, Cothran E Gus, McDonell Sue, Kenney Daniel G, Lear Teri L, Adelson David L, Chowdhary Bhanu P, Raudsepp Terje
Department of Veterinary Integrative Biosciences, College of Veterinary Medicine, Texas A&M University, College Station, Texas, United States of America.
School of Molecular and Biomedical Science, The University of Adelaide, Adelaide, South Australia, Australia.
PLoS Genet. 2014 Oct 23;10(10):e1004712. doi: 10.1371/journal.pgen.1004712. eCollection 2014 Oct.
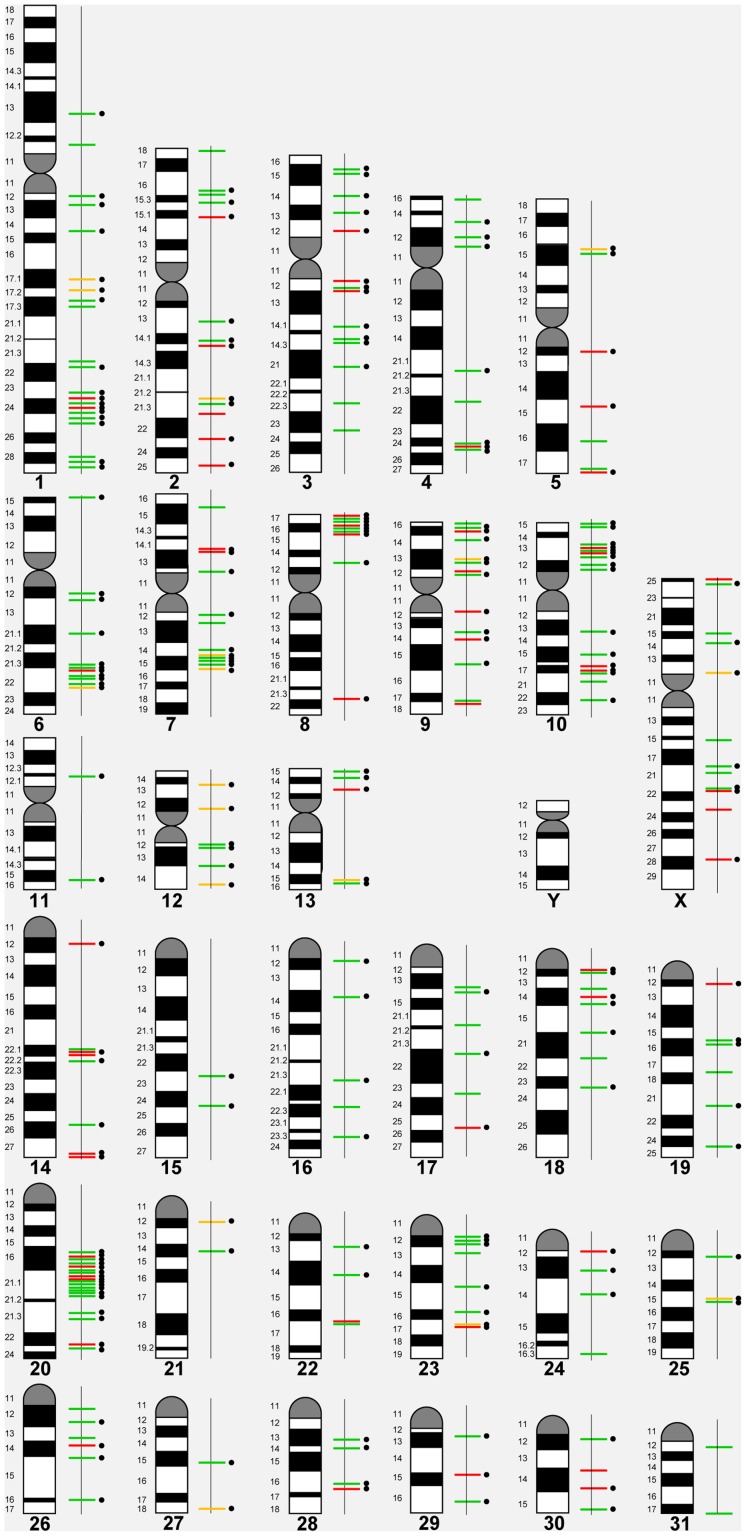
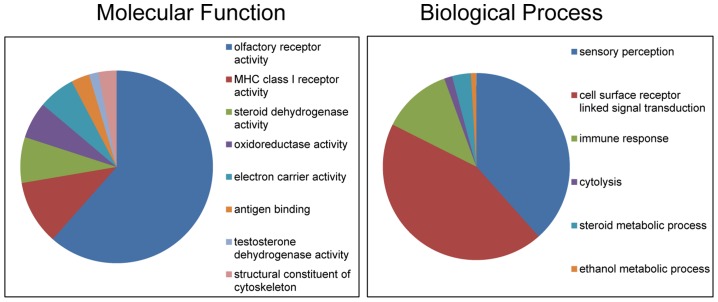
We constructed a 400K WG tiling oligoarray for the horse and applied it for the discovery of copy number variations (CNVs) in 38 normal horses of 16 diverse breeds, and the Przewalski horse. Probes on the array represented 18,763 autosomal and X-linked genes, and intergenic, sub-telomeric and chrY sequences. We identified 258 CNV regions (CNVRs) across all autosomes, chrX and chrUn, but not in chrY. CNVs comprised 1.3% of the horse genome with chr12 being most enriched. American Miniature horses had the highest and American Quarter Horses the lowest number of CNVs in relation to Thoroughbred reference. The Przewalski horse was similar to native ponies and draft breeds. The majority of CNVRs involved genes, while 20% were located in intergenic regions. Similar to previous studies in horses and other mammals, molecular functions of CNV-associated genes were predominantly in sensory perception, immunity and reproduction. The findings were integrated with previous studies to generate a composite genome-wide dataset of 1476 CNVRs. Of these, 301 CNVRs were shared between studies, while 1174 were novel and require further validation. Integrated data revealed that to date, 41 out of over 400 breeds of the domestic horse have been analyzed for CNVs, of which 11 new breeds were added in this study. Finally, the composite CNV dataset was applied in a pilot study for the discovery of CNVs in 6 horses with XY disorders of sexual development. A homozygous deletion involving AKR1C gene cluster in chr29 in two affected horses was considered possibly causative because of the known role of AKR1C genes in testicular androgen synthesis and sexual development. While the findings improve and integrate the knowledge of CNVs in horses, they also show that for effective discovery of variants of biomedical importance, more breeds and individuals need to be analyzed using comparable methodological approaches.
我们构建了一个用于马的400K全基因组平铺寡核苷酸阵列,并将其应用于16个不同品种的38匹正常马以及普氏野马中拷贝数变异(CNV)的发现。该阵列上的探针代表18,763个常染色体和X连锁基因,以及基因间、亚端粒和Y染色体序列。我们在所有常染色体、X染色体和未定位染色体(chrUn)上鉴定出258个CNV区域(CNVR),但在Y染色体上未发现。CNV占马基因组的1.3%,其中12号染色体最为富集。与纯血马参考基因组相比,美国迷你马的CNV数量最多,美国夸特马的CNV数量最少。普氏野马与本土矮种马和挽马品种相似。大多数CNVR涉及基因,而20%位于基因间区域。与之前对马和其他哺乳动物的研究相似,与CNV相关基因的分子功能主要集中在感官感知、免疫和繁殖方面。这些发现与之前的研究相结合,生成了一个包含1476个CNVR的全基因组综合数据集。其中,301个CNVR在不同研究中共享,而1174个是新发现的,需要进一步验证。综合数据显示,迄今为止,已对400多个家马品种中的41个进行了CNV分析,本研究新增了11个品种。最后,该综合CNV数据集被应用于一项初步研究,以发现6匹性发育XY障碍马中的CNV。由于已知AKR1C基因在睾丸雄激素合成和性发育中的作用,在两匹患病马中发现的29号染色体上涉及AKR1C基因簇的纯合缺失被认为可能是致病原因。虽然这些发现改进并整合了马中CNV的知识,但它们也表明,为了有效发现具有生物医学重要性的变异,需要使用可比的方法分析更多的品种和个体。