Schelleckes Michael, Lenders Malte, Guske Katrin, Schmitz Boris, Tanislav Christian, Ständer Sonja, Metze Dieter, Katona Istvan, Weis Joachim, Brand Stefan-Martin, Duning Thomas, Brand Eva
Internal Medicine D, Department of Nephrology, Hypertension and Rheumatology, University Hospital Muenster, Albert-Schweitzer-Campus 1, 48149, Muenster, Germany.
Institute of Sports Medicine, Molecular Genetics of Cardiovascular Disease, University Hospital Muenster, Horstmarer Landweg 39, 48149, Muenster, Germany.
Orphanet J Rare Dis. 2014 Nov 26;9:178. doi: 10.1186/s13023-014-0178-5.
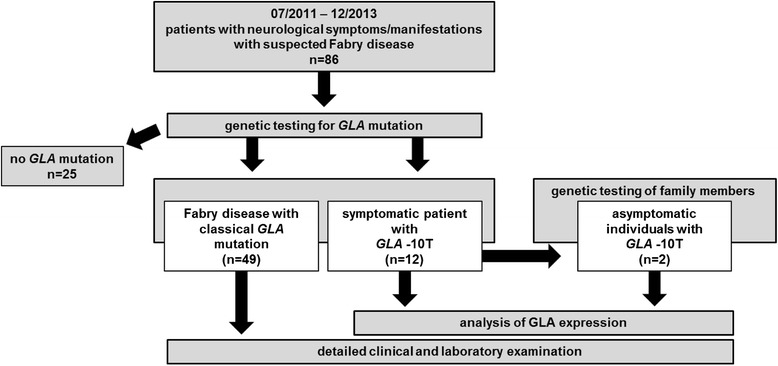
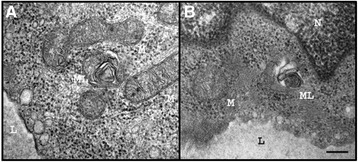
Fabry disease (FD) is a multisystemic disorder with typical neurological manifestations such as stroke and small fiber neuropathy (SFN), caused by mutations of the alpha-galactosidase A (GLA) gene. We analyzed 15 patients carrying the GLA haplotype -10C>T [rs2071225], IVS2-81_-77delCAGCC [rs5903184], IVS4-16A>G [rs2071397], and IVS6-22C>T [rs2071228] for potential neurological manifestations.
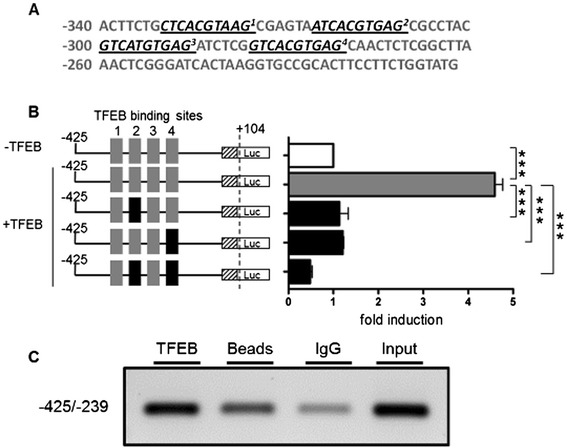
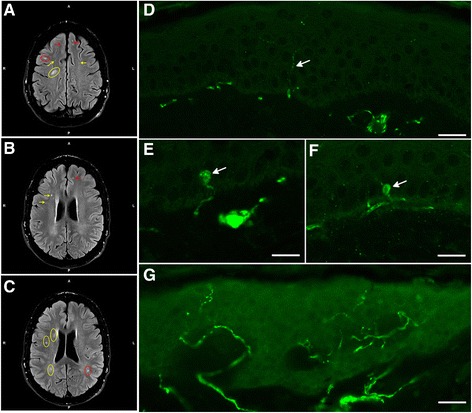
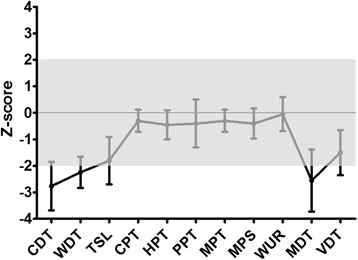
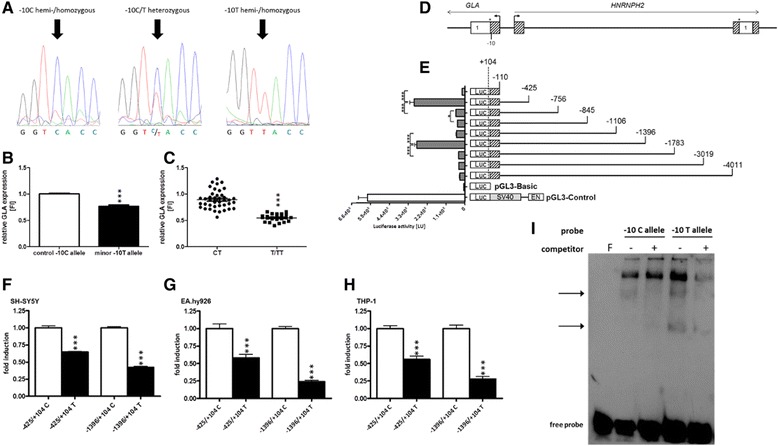
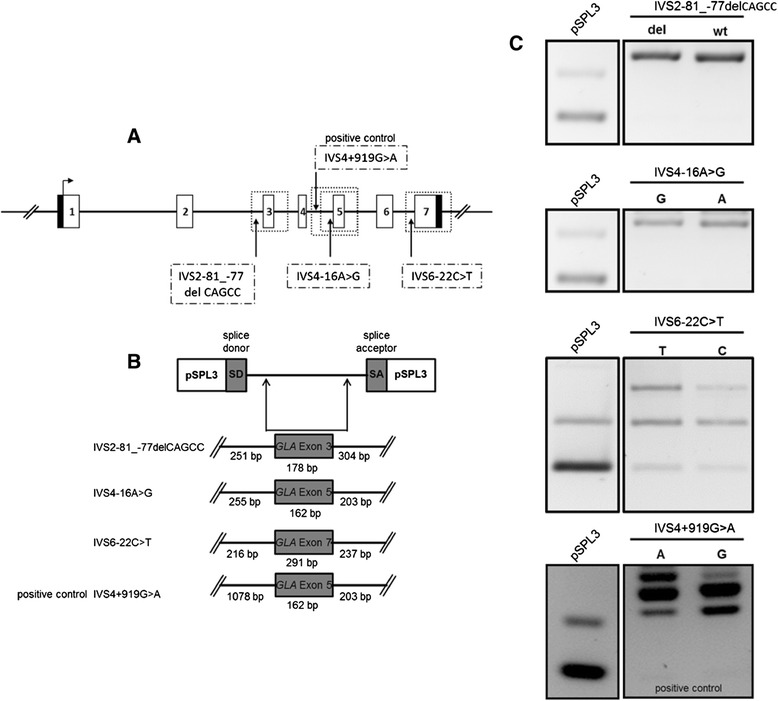
Patients were retrospectively analyzed for stroke, transient ischemic attack (TIA), white matter lesions (WML) and SFN with neuropathic pain. Functional impact of the haplotype was determined by molecular genetic methods including real-time PCR, exon trapping, promoter deletion constructs and electrophoretic mobility shift assays. Symptomatic -10T allele carriers suffered from stroke, TIA, WML, and SFN with neuropathic pain. Patients' mean GLA mRNA expression level was reduced to ~70% (p < 0.0001) and a dose-dependent effect of the -10T allele on GLA mRNA expression was observed in hemi/homozygous compared to heterozygous patients (p < 0.0001). Molecular analyzes revealed that the -10T allele resulted in a reduced promoter activity and an altered transcription factor binding, while a functional relevance of the co-segregated intronic variants was excluded by exon trapping.
Based on this complementary approach of clinical observation and functional testing, we conclude that the GLA -10T allele could be causal for the observed neurological manifestations. Future studies are needed to clarify whether affected patients benefit from GLA enzyme replacement therapy for end-organ damage prevention.
法布里病(FD)是一种多系统疾病,由α - 半乳糖苷酶A(GLA)基因突变引起,具有典型的神经学表现,如中风和小纤维神经病变(SFN)。我们分析了15名携带GLA单倍型-10C>T [rs2071225]、IVS2-81_-77delCAGCC [rs5903184]、IVS4-16A>G [rs2071397]和IVS6-22C>T [rs2071228]的患者的潜在神经学表现。
对患者进行回顾性分析,以评估中风、短暂性脑缺血发作(TIA)、白质病变(WML)和伴有神经性疼痛的SFN。通过包括实时PCR、外显子捕获、启动子缺失构建体和电泳迁移率变动分析在内的分子遗传学方法确定单倍型的功能影响。有症状的-10T等位基因携带者患有中风、TIA、WML和伴有神经性疼痛的SFN。患者的平均GLA mRNA表达水平降至约70%(p < 0.0001),与杂合子患者相比,在半合子/纯合子患者中观察到-10T等位基因对GLA mRNA表达的剂量依赖性效应(p < 0.0001)。分子分析显示,-10T等位基因导致启动子活性降低和转录因子结合改变,而通过外显子捕获排除了共分离内含子变体的功能相关性。
基于临床观察和功能测试的这种互补方法,我们得出结论,GLA -10T等位基因可能是观察到的神经学表现的病因。未来需要进行研究以阐明受影响的患者是否能从GLA酶替代疗法中受益以预防终末器官损伤。