Molecular Development of the Immune System Section, Laboratory of Immunology; NIAID Clinical Genomics Program; Human Immunological Diseases Unit, Laboratory of Host Defenses; and Intramural Clinical Management and Operations Branch, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD 20892 Molecular Development of the Immune System Section, Laboratory of Immunology; NIAID Clinical Genomics Program; Human Immunological Diseases Unit, Laboratory of Host Defenses; and Intramural Clinical Management and Operations Branch, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD 20892.
Department of Clinical Immunology, Children's Hospital of Fudan University, Shanghai 200433, China.
J Exp Med. 2014 Dec 15;211(13):2537-47. doi: 10.1084/jem.20141759. Epub 2014 Dec 8.
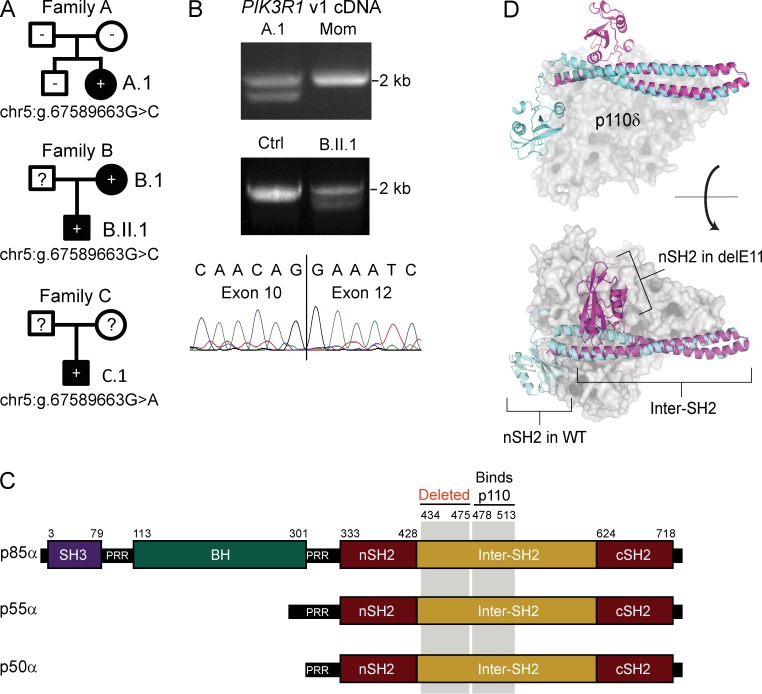
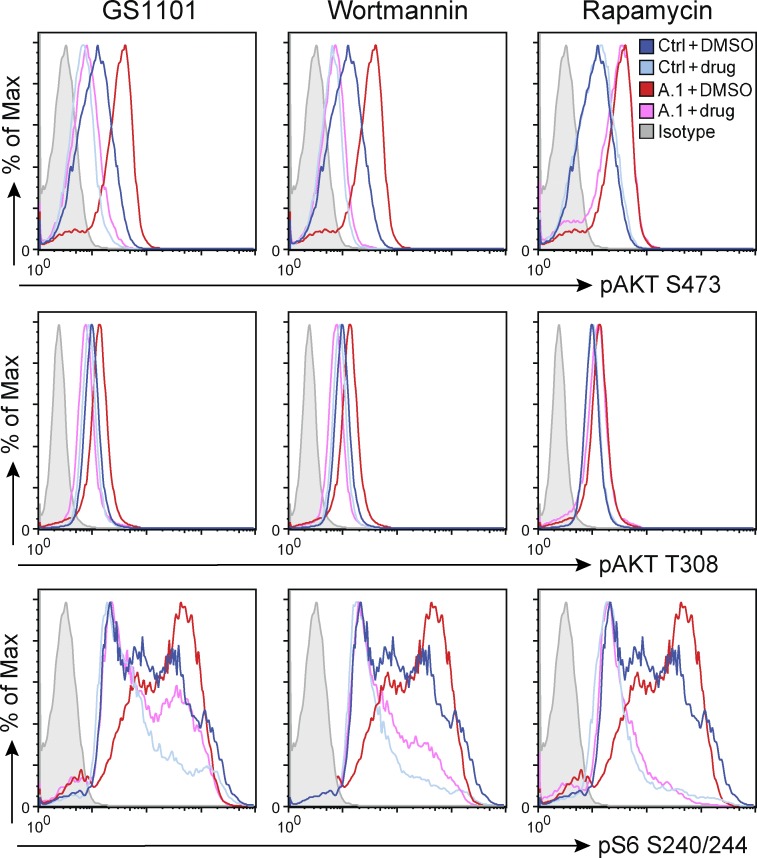
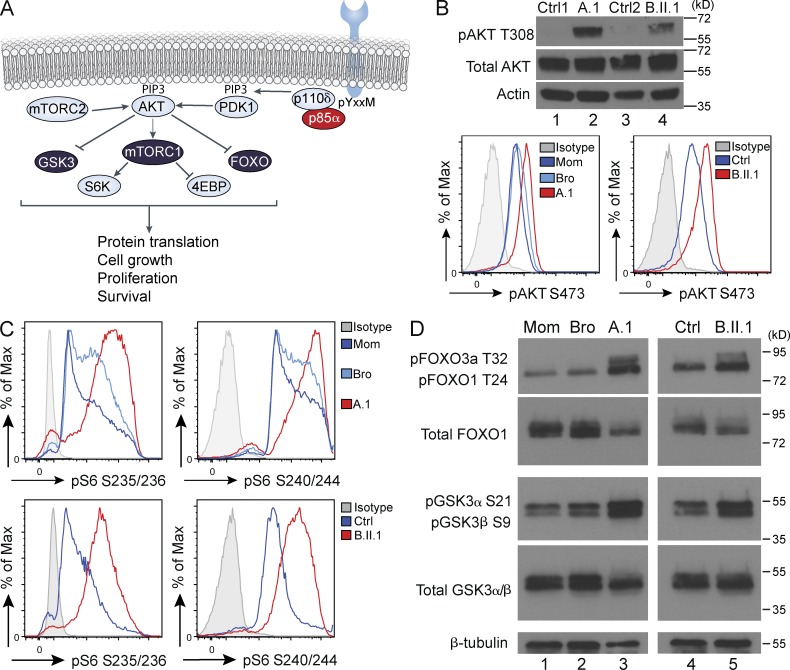
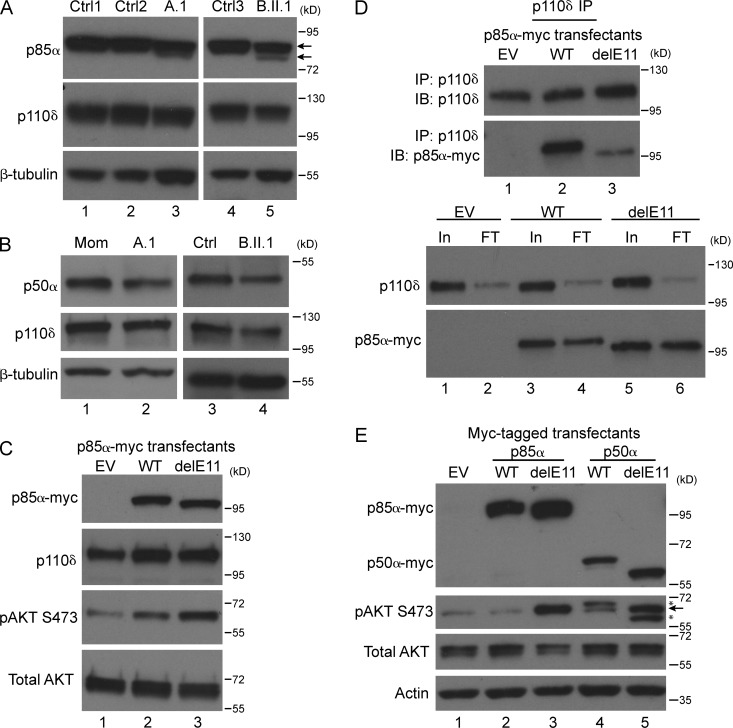
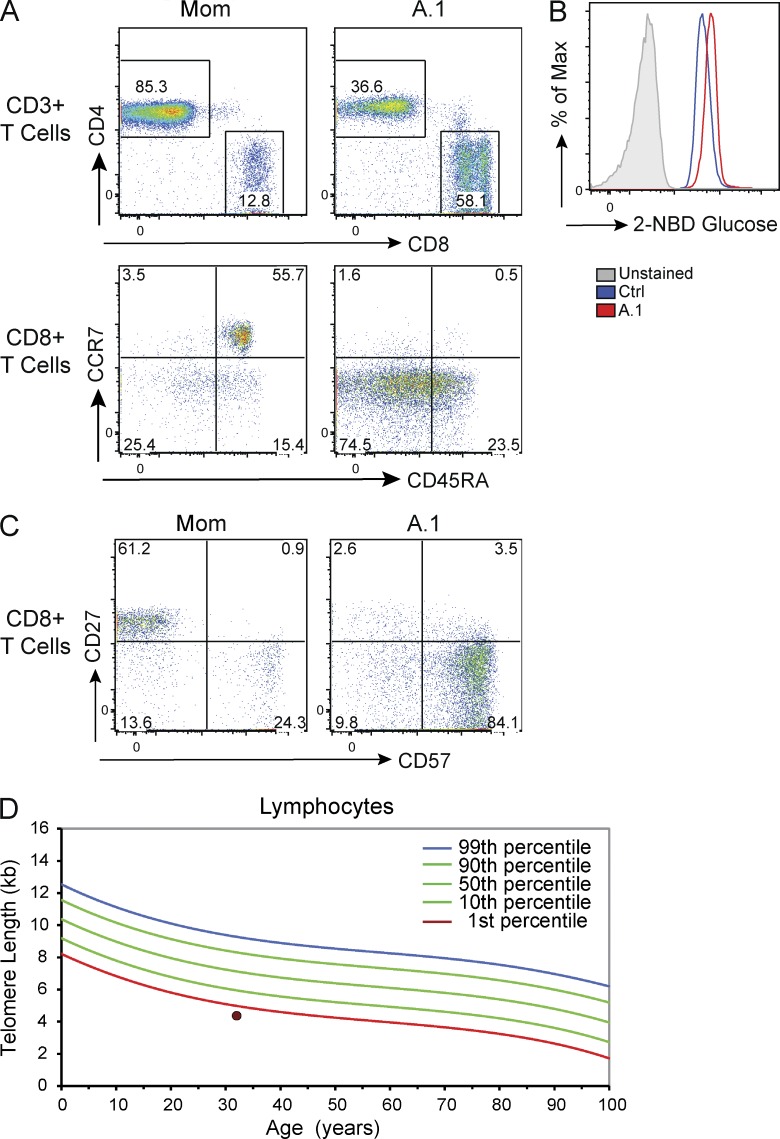
Class IA phosphatidylinositol 3-kinases (PI3K), which generate PIP3 as a signal for cell growth and proliferation, exist as an intracellular complex of a catalytic subunit bound to a regulatory subunit. We and others have previously reported that heterozygous mutations in PIK3CD encoding the p110δ catalytic PI3K subunit cause a unique disorder termed p110δ-activating mutations causing senescent T cells, lymphadenopathy, and immunodeficiency (PASLI) disease. We report four patients from three families with a similar disease who harbor a recently reported heterozygous splice site mutation in PIK3R1, which encodes the p85α, p55α, and p50α regulatory PI3K subunits. These patients suffer from recurrent sinopulmonary infections and lymphoproliferation, exhibit hyperactive PI3K signaling, and have prominent expansion and skewing of peripheral blood CD8(+) T cells toward terminally differentiated senescent effector cells with short telomeres. The PIK3R1 splice site mutation causes skipping of an exon, corresponding to loss of amino acid residues 434-475 in the inter-SH2 domain. The mutant p85α protein is expressed at low levels in patient cells and activates PI3K signaling when overexpressed in T cells from healthy subjects due to qualitative and quantitative binding changes in the p85α-p110δ complex and failure of the C-terminal region to properly inhibit p110δ catalytic activity.
IA 类磷脂酰肌醇 3-激酶 (PI3K) 作为细胞生长和增殖的信号,生成 PIP3,其存在于与调节亚基结合的催化亚基组成的细胞内复合物中。我们和其他人之前曾报道过,编码 p110δ 催化 PI3K 亚基的 PIK3CD 中的杂合突变导致一种独特的疾病,称为 p110δ 激活突变导致衰老 T 细胞、淋巴结病和免疫缺陷 (PASLI) 疾病。我们报告了来自三个家庭的四名患者患有类似疾病,他们携带最近报道的 PIK3R1 中的杂合剪接位点突变,该基因编码 p85α、p55α 和 p50α 调节 PI3K 亚基。这些患者患有复发性鼻窦和肺部感染以及淋巴增生,表现出过度活跃的 PI3K 信号,外周血 CD8(+) T 细胞明显扩增并向终末分化的衰老效应细胞(具有短端粒)倾斜。PIK3R1 剪接位点突变导致一个外显子跳过,对应于 SH2 结构域之间的氨基酸残基 434-475 的缺失。突变的 p85α 蛋白在患者细胞中表达水平较低,当在健康受试者的 T 细胞中过表达时,由于 p85α-p110δ 复合物的定性和定量结合变化以及 C 末端区域不能正确抑制 p110δ 催化活性,会激活 PI3K 信号。