Na Dokyun, Son Hyungbin, Gsponer Jörg
Department of Biochemistry and Molecular Biology, Centre for High-throughput Biology, University of British Columbia, 2125 East Mall, Vancouver, BC V6T 1Z4, Canada.
BMC Genomics. 2014 Dec 11;15(1):1091. doi: 10.1186/1471-2164-15-1091.
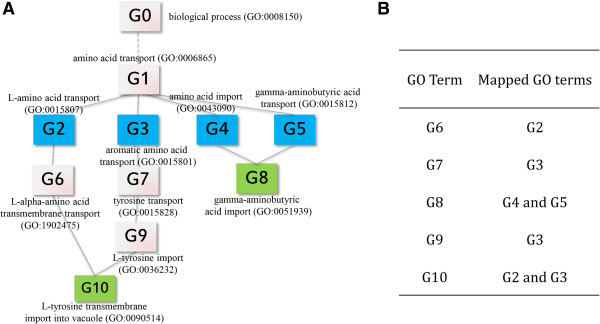
Communalities between large sets of genes obtained from high-throughput experiments are often identified by searching for enrichments of genes with the same Gene Ontology (GO) annotations. The GO analysis tools used for these enrichment analyses assume that GO terms are independent and the semantic distances between all parent-child terms are identical, which is not true in a biological sense. In addition these tools output lists of often redundant or too specific GO terms, which are difficult to interpret in the context of the biological question investigated by the user. Therefore, there is a demand for a robust and reliable method for gene categorization and enrichment analysis.
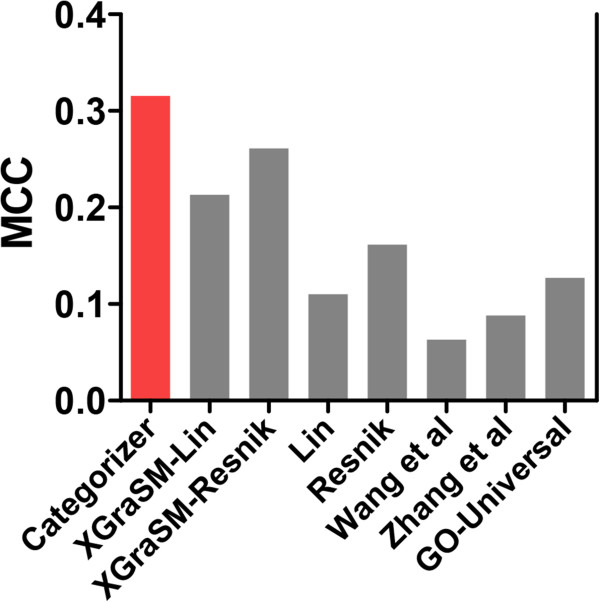
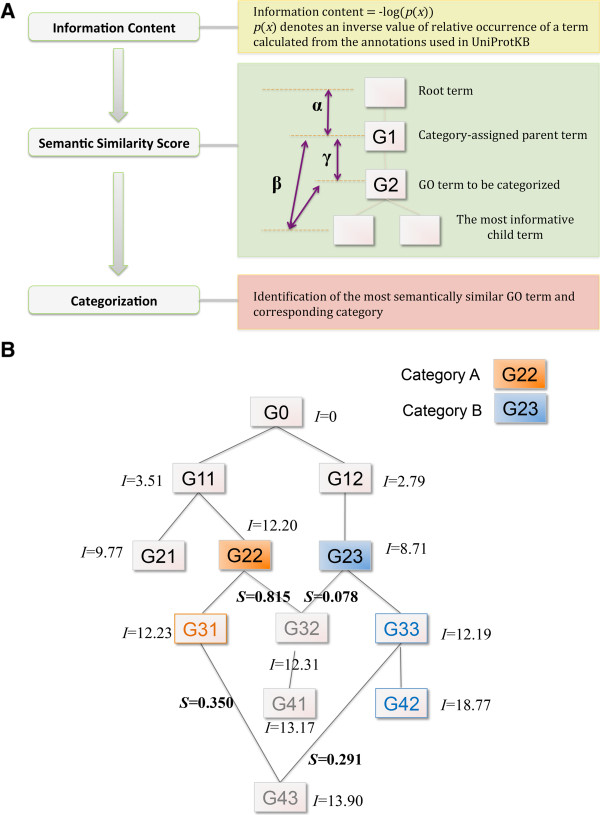
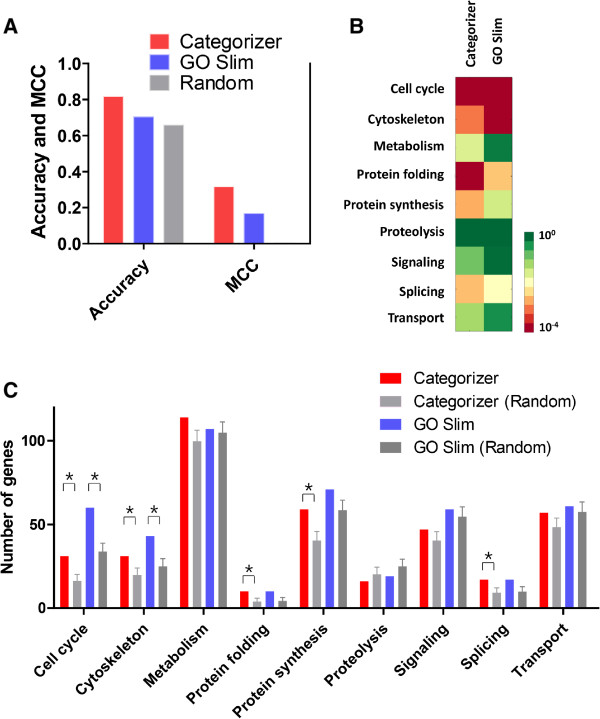
We have developed Categorizer, a tool that classifies genes into user-defined groups (categories) and calculates p-values for the enrichment of the categories. Categorizer identifies the biologically best-fit category for each gene by taking advantage of a specialized semantic similarity measure for GO terms. We demonstrate that Categorizer provides improved categorization and enrichment results of genetic modifiers of Huntington's disease compared to a classical GO Slim-based approach or categorizations using other semantic similarity measures.
Categorizer enables more accurate categorizations of genes than currently available methods. This new tool will help experimental and computational biologists analyzing genomic and proteomic data according to their specific needs in a more reliable manner.
从高通量实验中获得的大量基因之间的共性通常通过搜索具有相同基因本体论(GO)注释的基因富集情况来确定。用于这些富集分析的GO分析工具假定GO术语是独立的,并且所有父子术语之间的语义距离是相同的,而这在生物学意义上并不成立。此外,这些工具输出的往往是冗余或过于具体的GO术语列表,在用户所研究的生物学问题背景下很难解释。因此,需要一种强大且可靠的基因分类和富集分析方法。
我们开发了Categorizer工具,它可以将基因分类到用户定义的组(类别)中,并计算类别的富集p值。Categorizer通过利用一种专门的GO术语语义相似性度量,为每个基因确定生物学上最匹配的类别。我们证明,与基于经典GO Slim的方法或使用其他语义相似性度量的分类方法相比,Categorizer在亨廷顿舞蹈病基因修饰因子的分类和富集结果方面有改进。
与现有方法相比,Categorizer能够对基因进行更准确的分类。这个新工具将帮助实验生物学家和计算生物学家根据他们的特定需求,以更可靠的方式分析基因组和蛋白质组数据。