Maile Tobias M, Izrael-Tomasevic Anita, Cheung Tommy, Guler Gulfem D, Tindell Charles, Masselot Alexandre, Liang Jun, Zhao Feng, Trojer Patrick, Classon Marie, Arnott David
From the ‡Protein Chemistry Department, Genentech Inc., South San Francisco, California 94080;
§Cancer Targets Department, Genentech, Inc., South San Francisco, California 94080;
Mol Cell Proteomics. 2015 Apr;14(4):1148-58. doi: 10.1074/mcp.O114.046573. Epub 2015 Feb 13.
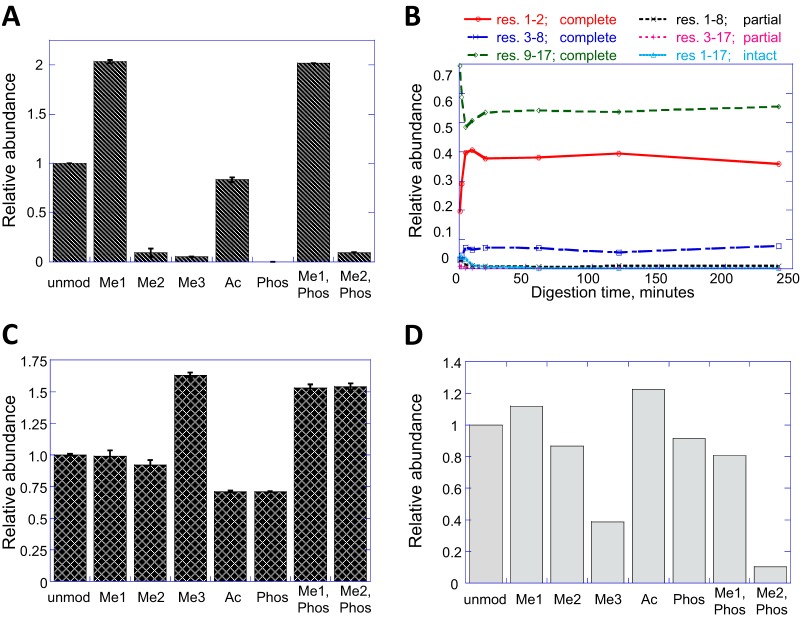
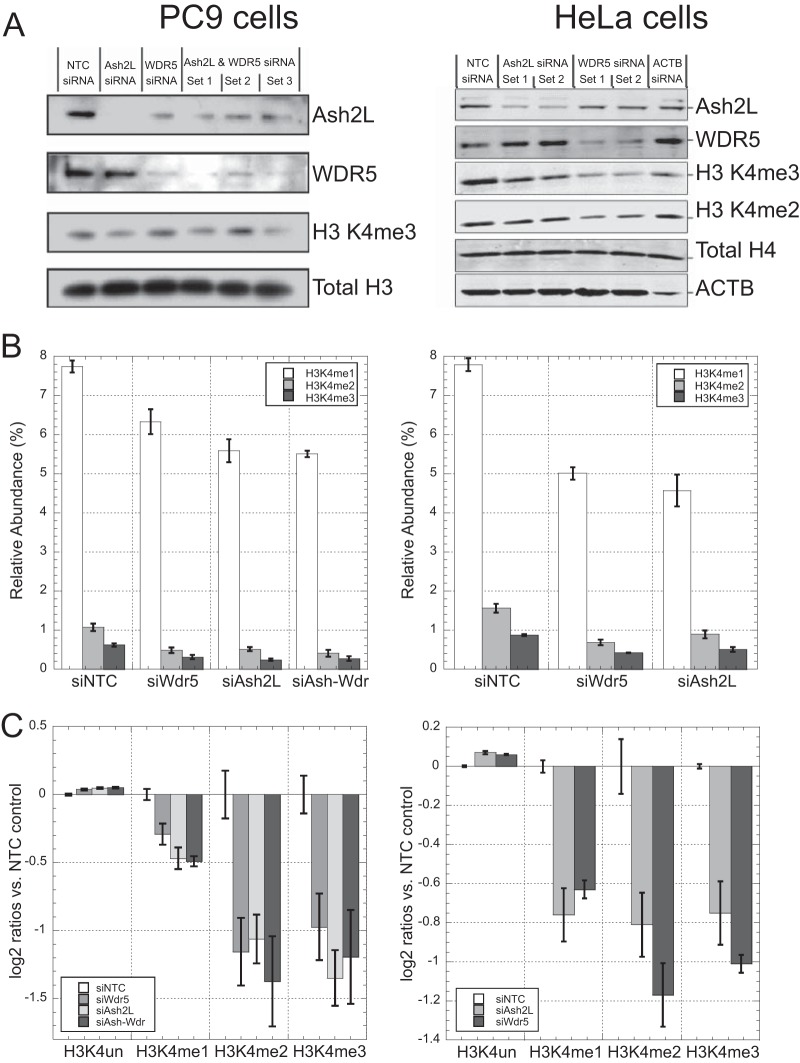
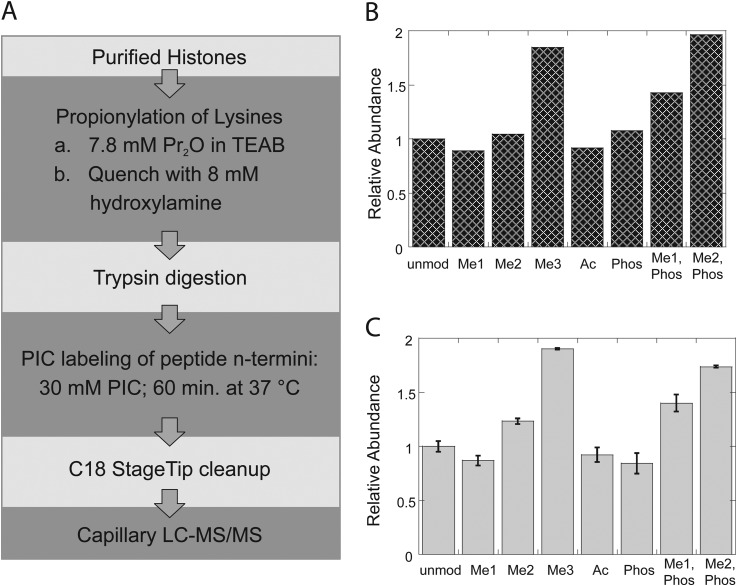
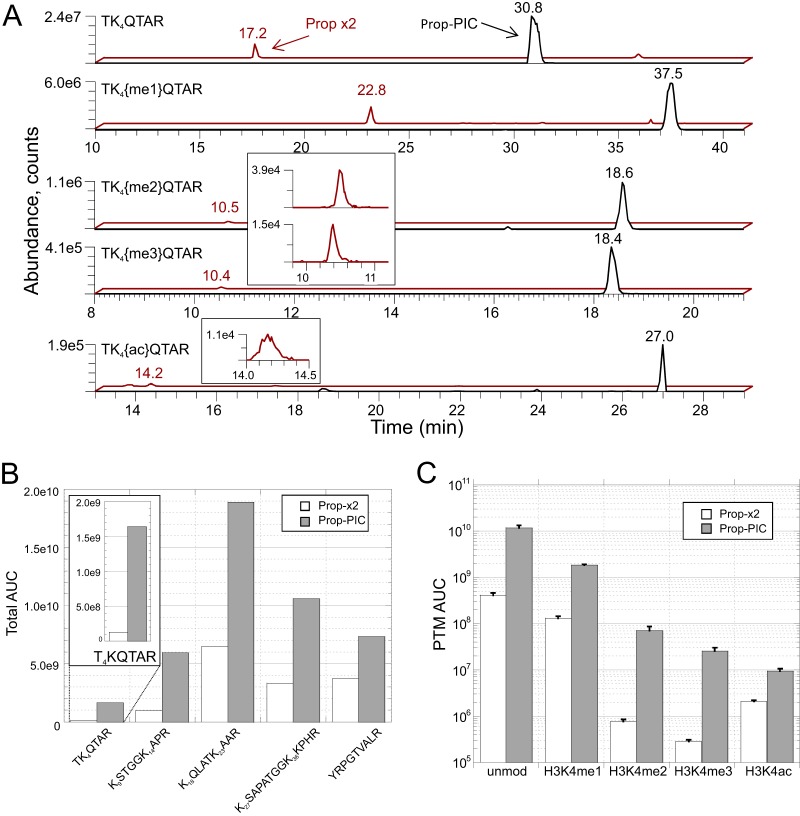
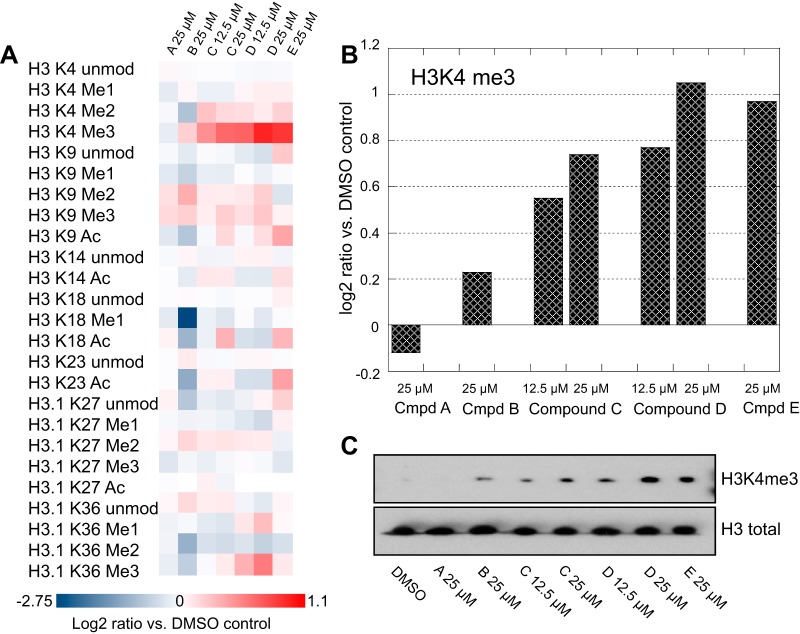
Mass spectrometry is a powerful alternative to antibody-based methods for the analysis of histone post-translational modifications (marks). A key development in this approach was the deliberate propionylation of histones to improve sequence coverage across the lysine-rich and hydrophilic tails that bear most modifications. Several marks continue to be problematic however, particularly di- and tri-methylated lysine 4 of histone H3 which we found to be subject to substantial and selective losses during sample preparation and liquid chromatography-mass spectrometry. We developed a new method employing a "one-pot" hybrid chemical derivatization of histones, whereby an initial conversion of free lysines to their propionylated forms under mild aqueous conditions is followed by trypsin digestion and labeling of new peptide N termini with phenyl isocyanate. High resolution mass spectrometry was used to collect qualitative and quantitative data, and a novel web-based software application (Fishtones) was developed for viewing and quantifying histone marks in the resulting data sets. Recoveries of 53 methyl, acetyl, and phosphoryl marks on histone H3.1 were improved by an average of threefold overall, and over 50-fold for H3K4 di- and tri-methyl marks. The power of this workflow for epigenetic research and drug discovery was demonstrated by measuring quantitative changes in H3K4 trimethylation induced by small molecule inhibitors of lysine demethylases and siRNA knockdown of epigenetic modifiers ASH2L and WDR5.
质谱分析法是一种强大的替代基于抗体的方法,用于分析组蛋白的翻译后修饰(标记)。这种方法的一个关键进展是对组蛋白进行有意的丙酰化,以提高对富含赖氨酸且带有大多数修饰的亲水性尾部的序列覆盖。然而,仍有几种标记存在问题,特别是组蛋白H3的赖氨酸4的二甲基化和三甲基化,我们发现其在样品制备和液相色谱 - 质谱分析过程中会大量且选择性地丢失。我们开发了一种新方法,采用组蛋白的“一锅法”混合化学衍生化,即在温和的水性条件下将游离赖氨酸初步转化为其丙酰化形式,然后进行胰蛋白酶消化并用异氰酸苯酯标记新的肽N末端。使用高分辨率质谱法收集定性和定量数据,并开发了一种基于网络的新型软件应用程序(Fishtones),用于查看和量化所得数据集中的组蛋白标记。组蛋白H3.1上53种甲基、乙酰基和磷酰基标记的回收率总体平均提高了三倍,H3K4二甲基和三甲基标记的回收率提高了50倍以上。通过测量赖氨酸去甲基化酶小分子抑制剂以及表观遗传修饰因子ASH2L和WDR5的siRNA敲低诱导的H3K4三甲基化的定量变化,证明了该工作流程在表观遗传学研究和药物发现中的作用。