Wittig Michael, Anmarkrud Jarl A, Kässens Jan C, Koch Simon, Forster Michael, Ellinghaus Eva, Hov Johannes R, Sauer Sascha, Schimmler Manfred, Ziemann Malte, Görg Siegfried, Jacob Frank, Karlsen Tom H, Franke Andre
Christian-Albrechts-University of Kiel, Institute of Clinical Molecular Biology, Kiel, Germany
Norwegian PSC Research Center, Department of Transplantation Medicine, Division of Cancer Medicine, Surgery and Transplantation, Oslo University Hospital, Rikshospitalet, Oslo, Norway K.G. Jebsen Inflammation Research Center, Institute of Clinical Medicine, University of Oslo, Oslo, Norway Research Institute of Internal Medicine, Division of Cancer Medicine, Surgery and Transplantation, Oslo University Hospital, Oslo, Norway.
Nucleic Acids Res. 2015 Jun 23;43(11):e70. doi: 10.1093/nar/gkv184. Epub 2015 Mar 9.
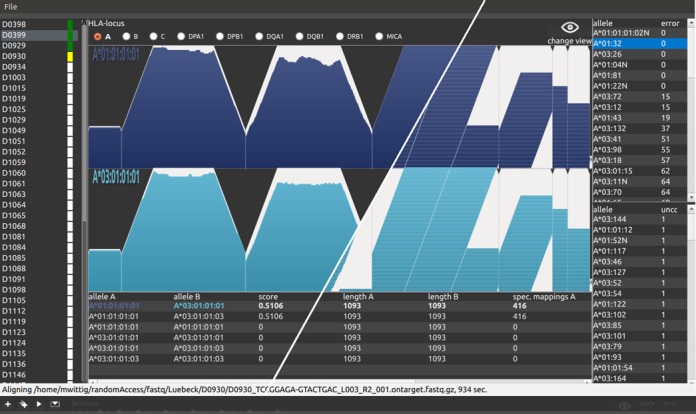
The human leukocyte antigen (HLA) complex contains the most polymorphic genes in the human genome. The classical HLA class I and II genes define the specificity of adaptive immune responses. Genetic variation at the HLA genes is associated with susceptibility to autoimmune and infectious diseases and plays a major role in transplantation medicine and immunology. Currently, the HLA genes are characterized using Sanger- or next-generation sequencing (NGS) of a limited amplicon repertoire or labeled oligonucleotides for allele-specific sequences. High-quality NGS-based methods are in proprietary use and not publicly available. Here, we introduce the first highly automated open-kit/open-source HLA-typing method for NGS. The method employs in-solution targeted capturing of the classical class I (HLA-A, HLA-B, HLA-C) and class II HLA genes (HLA-DRB1, HLA-DQA1, HLA-DQB1, HLA-DPA1, HLA-DPB1). The calling algorithm allows for highly confident allele-calling to three-field resolution (cDNA nucleotide variants). The method was validated on 357 commercially available DNA samples with known HLA alleles obtained by classical typing. Our results showed on average an accurate allele call rate of 0.99 in a fully automated manner, identifying also errors in the reference data. Finally, our method provides the flexibility to add further enrichment target regions.
人类白细胞抗原(HLA)复合体包含人类基因组中多态性最高的基因。经典的HLA I类和II类基因决定了适应性免疫反应的特异性。HLA基因的遗传变异与自身免疫性疾病和感染性疾病的易感性相关,并且在移植医学和免疫学中发挥着重要作用。目前,HLA基因是通过对有限扩增子文库进行桑格测序或下一代测序(NGS),或使用标记的寡核苷酸针对等位基因特异性序列来进行分型的。基于高质量NGS的方法属于专利技术,尚未公开。在此,我们介绍了第一种用于NGS的高度自动化的开放式试剂盒/开源HLA分型方法。该方法采用溶液内靶向捕获经典的I类(HLA-A、HLA-B、HLA-C)和II类HLA基因(HLA-DRB1、HLA-DQA1、HLA-DQB1、HLA-DPA1、HLA-DPB1)。分型算法允许以高置信度将等位基因分型到三位分辨率(cDNA核苷酸变体)。该方法在357个通过经典分型获得的已知HLA等位基因的市售DNA样本上进行了验证。我们的结果显示,以全自动方式平均等位基因分型准确率为0.99,同时还识别出了参考数据中的错误。最后,我们的方法提供了添加更多富集目标区域的灵活性。