Maroja Luana S, Larson Erica L, Bogdanowicz Steven M, Harrison Richard G
Department of Biology, Williams College, Williamstown, Massachusetts 01267
Division of Biological Sciences, University of Montana, Missoula, Montana 59812.
G3 (Bethesda). 2015 Aug 26;5(11):2219-27. doi: 10.1534/g3.115.021246.
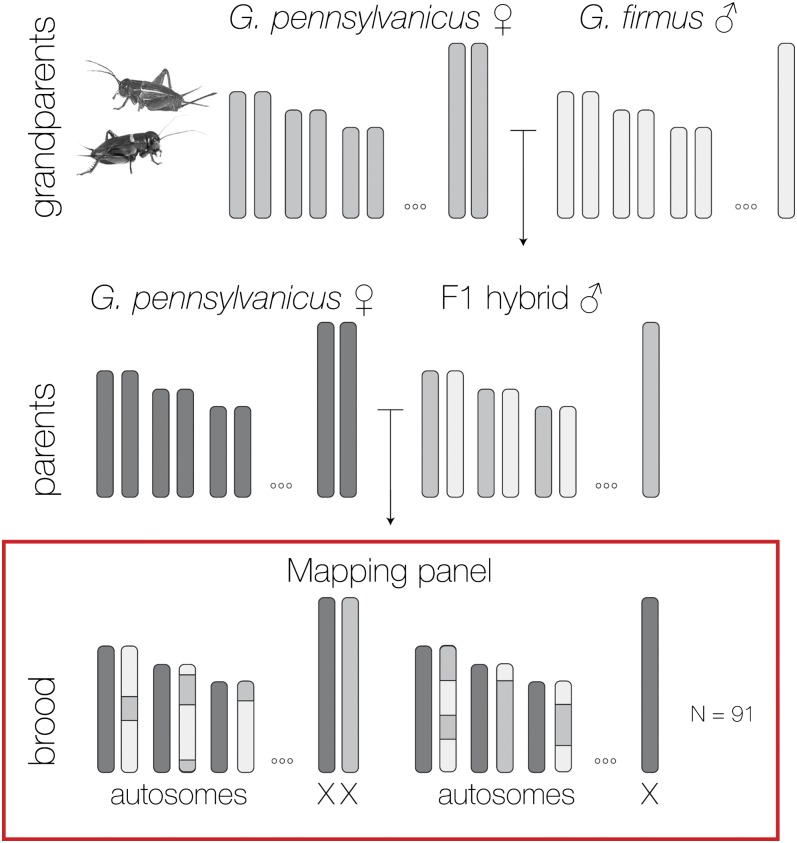
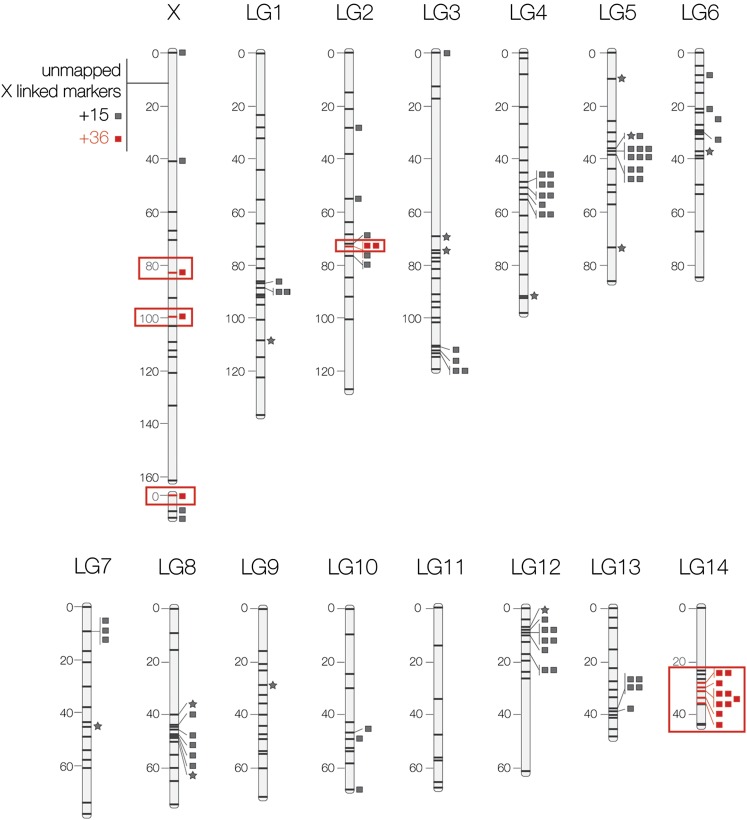
Characterizing the extent of genomic differentiation between recently diverged lineages provides an important context for understanding the early stages of speciation. When such lineages form discrete hybrid zones, patterns of differential introgression allow direct estimates of which genome regions are likely involved in speciation and local adaptation. Here we use a backcross experimental design to construct a genetic linkage map for the field crickets Gryllus firmus and Gryllus pennsylvanicus, which interact in a well-characterized hybrid zone in eastern North America. We demonstrate that loci with major allele frequency differences between allopatric populations are not randomly distributed across the genome. Instead, most are either X-linked or map to a few small autosomal regions. Furthermore, the subset of those highly differentiated markers that exhibit restricted introgression across the cricket hybrid zone are also concentrated on the X chromosome (39 of 50 loci) and in a single 7-cM region of one autosome. Although the accumulation on the sex chromosome of genes responsible for postzygotic barriers is a well-known phenomenon, less attention has been given to the genomic distribution of genes responsible for prezygotic barriers. We discuss the implications of our results for speciation, both in the context of the role of sex chromosomes and also with respect to the likely causes of heterogeneous genomic divergence. Although we do not yet have direct evidence for the accumulation of ecological, behavioral, or fertilization prezygotic barrier genes on the X chromosome, faster-X evolution could make these barriers more likely to be X-linked.
描绘最近分化的谱系之间基因组分化的程度,为理解物种形成的早期阶段提供了重要背景。当这些谱系形成离散的杂交带时,差异渗入模式可以直接估计哪些基因组区域可能参与物种形成和局部适应。在这里,我们采用回交实验设计,为北美东部一个特征明确的杂交带中相互作用的田野蟋蟀(Gryllus firmus)和宾夕法尼亚蟋蟀(Gryllus pennsylvanicus)构建遗传连锁图谱。我们证明,在异域种群之间具有主要等位基因频率差异的位点并非随机分布在基因组中。相反,大多数位点要么位于X染色体上,要么映射到少数几个小的常染色体区域。此外,那些在蟋蟀杂交带中表现出有限渗入的高度分化标记的子集也集中在X染色体上(50个位点中的39个)和一条常染色体的一个单一的7厘摩区域。虽然负责合子后屏障的基因在性染色体上的积累是一个众所周知的现象,但对于负责合子前屏障的基因的基因组分布关注较少。我们讨论了我们的结果对物种形成的影响,既从性染色体的作用方面,也从基因组异质分化的可能原因方面。虽然我们尚未有直接证据表明生态、行为或受精合子前屏障基因在X染色体上积累,但X染色体的快速进化可能使这些屏障更有可能与X染色体连锁。