Katoh-Fukui Yuko, Igarashi Maki, Nagasaki Keisuke, Horikawa Reiko, Nagai Toshiro, Tsuchiya Takayoshi, Suzuki Erina, Miyado Mami, Hata Kenichiro, Nakabayashi Kazuhiko, Hayashi Keiko, Matsubara Yoichi, Baba Takashi, Morohashi Ken-Ichirou, Igarashi Arisa, Ogata Tsutomu, Takada Shuji, Fukami Maki
Department of Molecular Endocrinology National Research Institute for Child Health and Development Tokyo Japan.
Division of Pediatrics Department of Homeostatic Regulation and Development Niigata University Graduate School of Medical and Dental Sciences Niigata Japan.
Mol Genet Genomic Med. 2015 Jul 14;3(6):550-7. doi: 10.1002/mgg3.165. eCollection 2015 Nov.
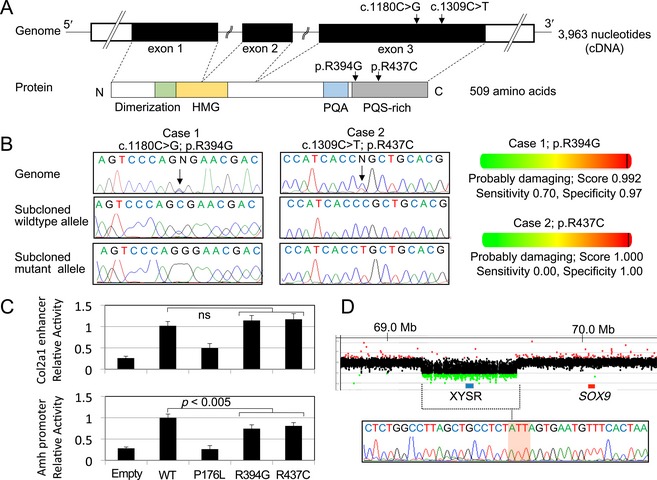
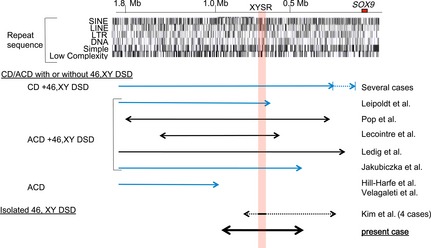
SOX9 haploinsufficiency underlies campomelic dysplasia (CD) with or without testicular dysgenesis. Current understanding of the phenotypic variability and mutation spectrum of SOX9 abnormalities remains fragmentary. Here, we report three patients with hitherto unreported SOX9 abnormalities. These patients were identified through molecular analysis of 33 patients with 46,XY disorders of sex development (DSD). Patients 1-3 manifested testicular dysgenesis or regression without CD. Patients 1 and 2 carried probable damaging mutations p.Arg394Gly and p.Arg437Cys, respectively, in the SOX9 C-terminal domain but not in other known 46,XY DSD causative genes. These substitutions were absent from ~120,000 alleles in the exome database. These mutations retained normal transactivating activity for the Col2a1 enhancer, but showed impaired activity for the Amh promoter. Patient 3 harbored a maternally inherited ~491 kb SOX9 upstream deletion that encompassed the known 32.5 kb XY sex reversal region. Breakpoints of the deletion resided within nonrepeat sequences and were accompanied by a short-nucleotide insertion. The results imply that testicular dysgenesis and regression without skeletal dysplasia may be rare manifestations of SOX9 abnormalities. Furthermore, our data broaden pathogenic SOX9 abnormalities to include C-terminal missense substitutions which lead to target-gene-specific protein dysfunction, and enhancer-containing upstream microdeletions mediated by nonhomologous end-joining.
SOX9单倍体不足是导致有或无睾丸发育不全的弯肢侏儒症(CD)的原因。目前对SOX9异常的表型变异性和突变谱的理解仍然不完整。在此,我们报告三例具有迄今未报道的SOX9异常的患者。这些患者是通过对33例46,XY性发育障碍(DSD)患者进行分子分析而确定的。患者1-3表现为无CD的睾丸发育不全或退化。患者1和2在SOX9 C末端结构域分别携带可能具有损害性的突变p.Arg394Gly和p.Arg437Cys,但在其他已知的46,XY DSD致病基因中未发现。外显子数据库中约120,000个等位基因中不存在这些替代。这些突变对Col2a1增强子保留正常的反式激活活性,但对Amh启动子显示活性受损。患者3携带一个母系遗传的约491 kb的SOX9上游缺失,该缺失包含已知的32.5 kb XY性反转区域。缺失的断点位于非重复序列内,并伴有短核苷酸插入。结果表明,无骨骼发育异常的睾丸发育不全和退化可能是SOX9异常的罕见表现。此外,我们的数据将致病性SOX9异常扩大到包括导致靶基因特异性蛋白质功能障碍的C末端错义替代以及由非同源末端连接介导的含增强子的上游微缺失。