Institute of Biochemistry, Medical Faculty, University of Giessen, Friedrichstrasse 24, 35392 Giessen, Germany.
Department of Chemistry, University of Eastern Finland, PO BOX 111, 80101 Joensuu, Finland.
Sci Rep. 2016 Nov 23;6:37583. doi: 10.1038/srep37583.
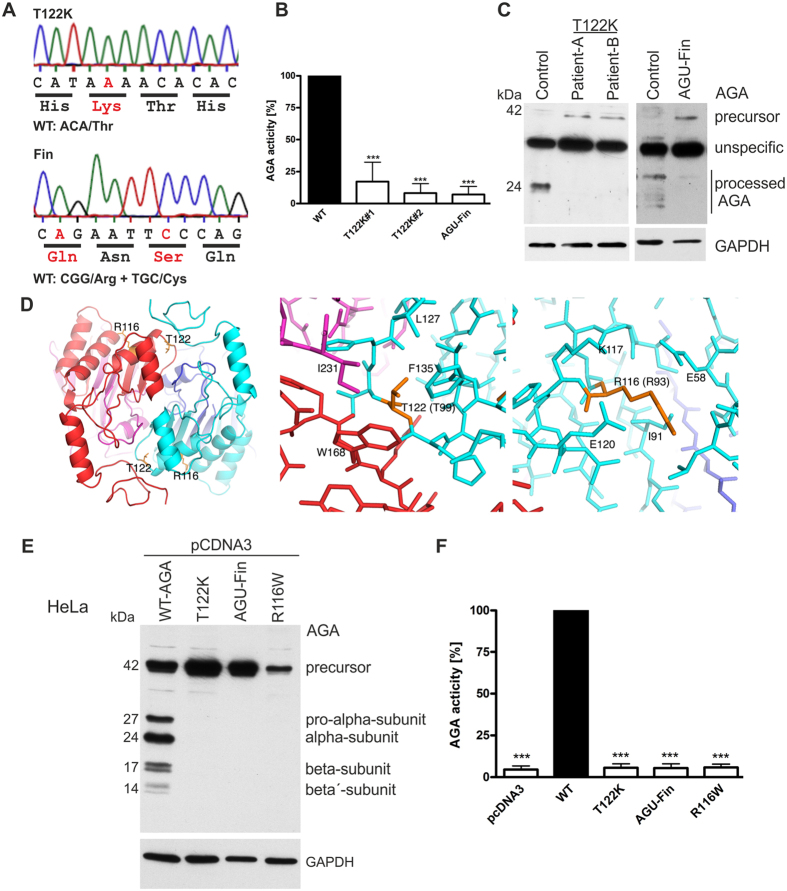
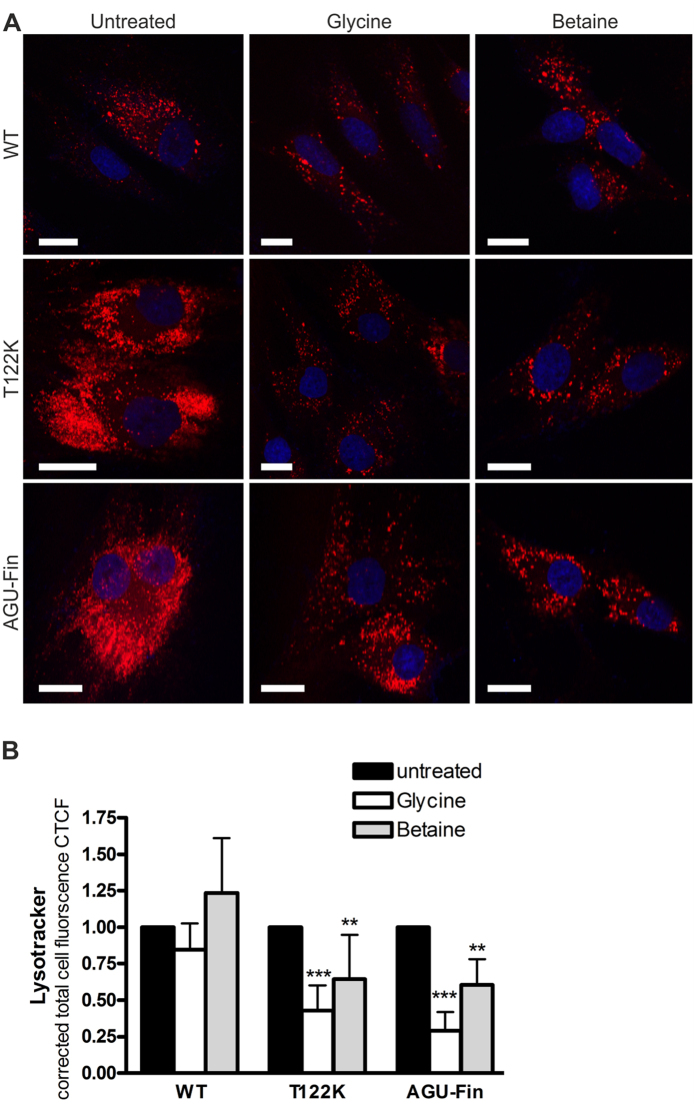
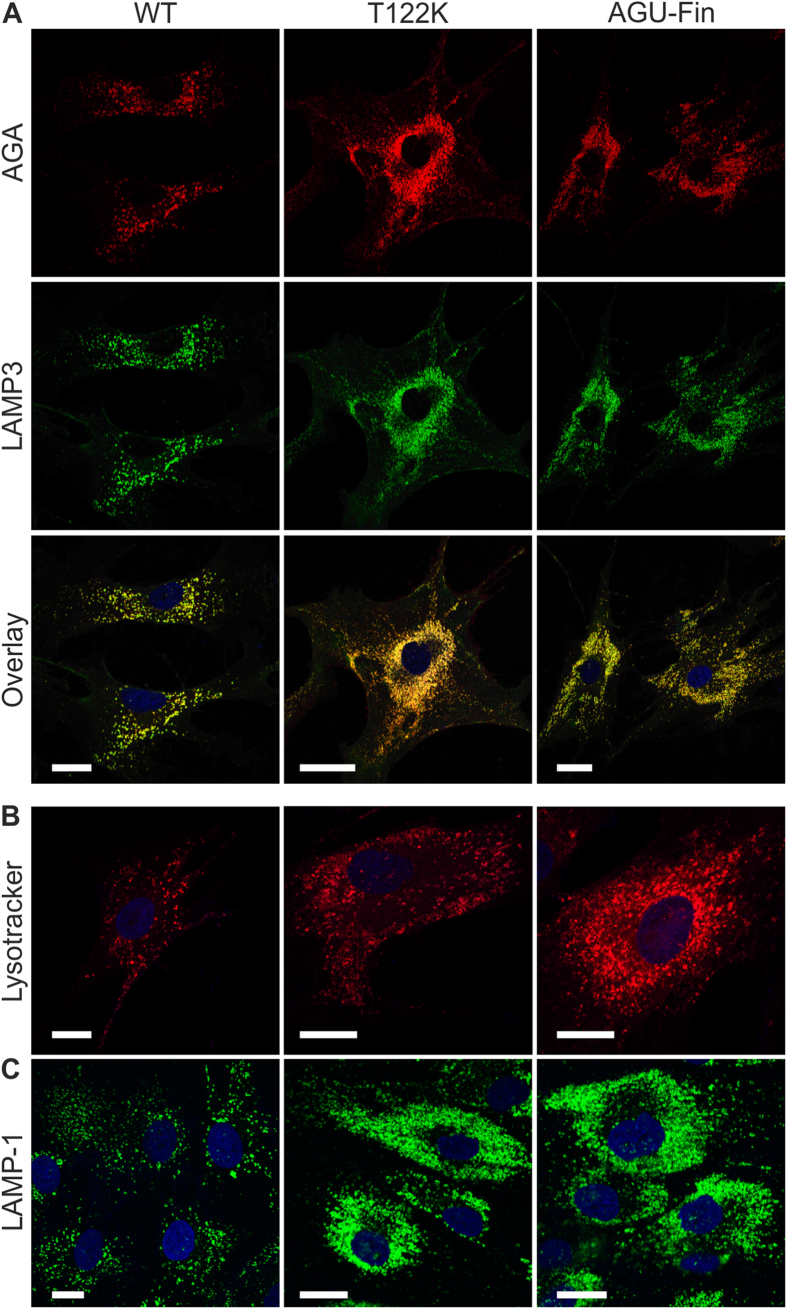
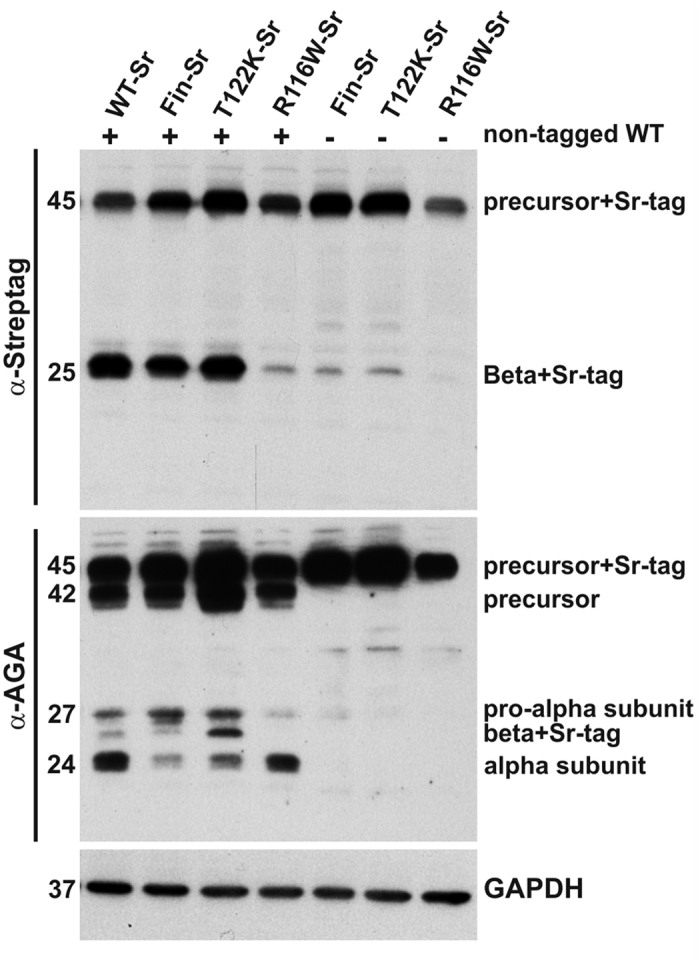
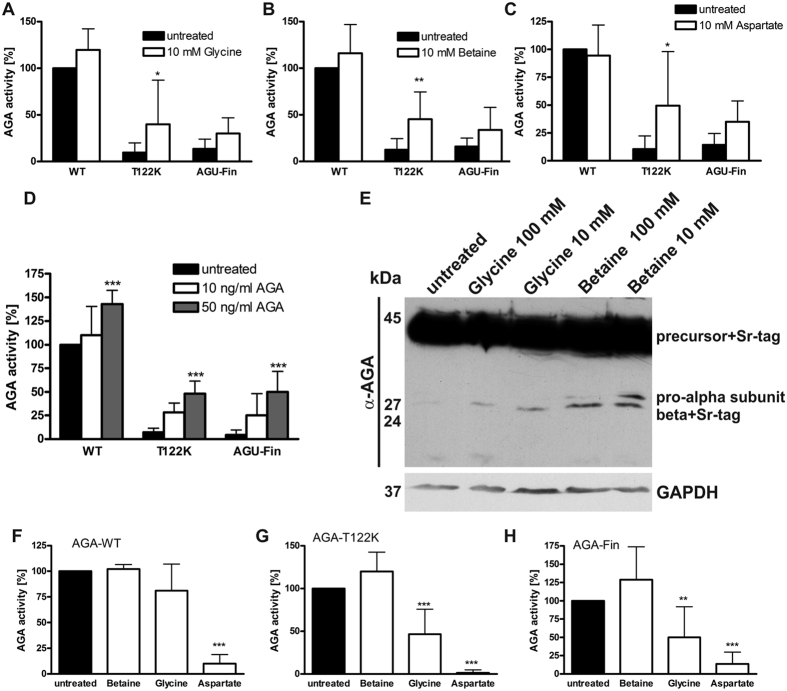
Aspartylglucosaminuria (AGU) is a lysosomal storage disorder that is caused by genetic deficiency of the enzyme aspartylglucosaminidase (AGA) which is involved in glycoprotein degradation. AGU is a progressive disorder that results in severe mental retardation in early adulthood. No curative therapy is currently available for AGU. We have here characterized the consequences of a novel AGU mutation that results in Thr122Lys exchange in AGA, and compared this mutant form to one carrying the worldwide most common AGU mutation, AGU-Fin. We show that T122K mutated AGA is expressed in normal amounts and localized in lysosomes, but exhibits low AGA activity due to impaired processing of the precursor molecule into subunits. Coexpression of T122K with wildtype AGA results in processing of the precursor into subunits, implicating that the mutation causes a local misfolding that prevents the precursor from becoming processed. Similar data were obtained for the AGU-Fin mutant polypeptide. We have here also identified small chemical compounds that function as chemical or pharmacological chaperones for the mutant AGA. Treatment of patient fibroblasts with these compounds results in increased AGA activity and processing, implicating that these substances may be suitable for chaperone mediated therapy for AGU.
天冬氨酰葡糖胺尿症(AGU)是一种溶酶体贮积病,由天冬氨酰葡糖胺酶(AGA)的遗传缺陷引起,该酶参与糖蛋白降解。AGU 是一种进行性疾病,导致成年早期严重智力迟钝。目前尚无针对 AGU 的治愈疗法。我们在这里描述了 AGA 中 Thr122Lys 交换导致的新型 AGU 突变的后果,并将这种突变形式与携带最常见的 AGU 突变的 AGA-Fin 进行了比较。我们表明,T122K 突变的 AGA 以正常量表达并定位于溶酶体中,但由于前体分子转化为亚基的过程受损,表现出低 AGA 活性。T122K 与野生型 AGA 的共表达导致前体转化为亚基,表明该突变导致局部错误折叠,阻止前体被加工。AGU-Fin 突变多肽也获得了类似的数据。我们还在这里鉴定了一些小分子化合物,它们可作为突变 AGA 的化学或药理学伴侣。用这些化合物处理患者成纤维细胞可增加 AGA 活性和加工,表明这些物质可能适合用于 AGU 的伴侣介导治疗。