Gruber Robert, Rogerson Clare, Windpassinger Christian, Banushi Blerida, Straatman-Iwanowska Anna, Hanley Joanna, Forneris Federico, Strohal Robert, Ulz Peter, Crumrine Debra, Menon Gopinathan K, Blunder Stefan, Schmuth Matthias, Müller Thomas, Smith Holly, Mills Kevin, Kroisel Peter, Janecke Andreas R, Gissen Paul
Department of Dermatology, Medical University of Innsbruck, Innsbruck, Austria; Division of Human Genetics, Medical University of Innsbruck, Innsbruck, Austria.
MRC Laboratory for Molecular Cell Biology, University College London, London, UK; Institute of Child Health, University College London, London, UK.
J Invest Dermatol. 2017 Apr;137(4):845-854. doi: 10.1016/j.jid.2016.12.010. Epub 2016 Dec 23.
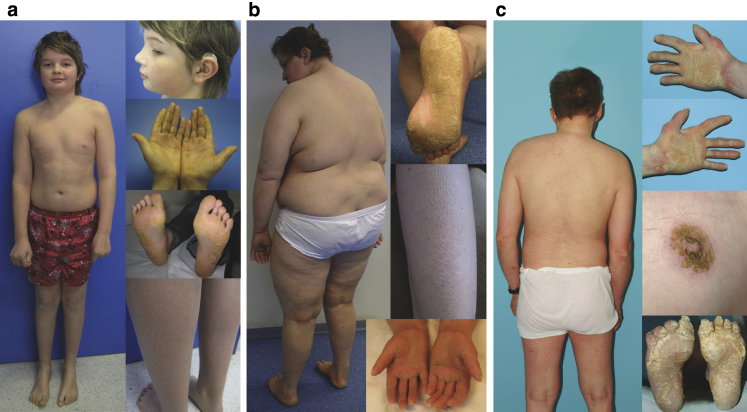
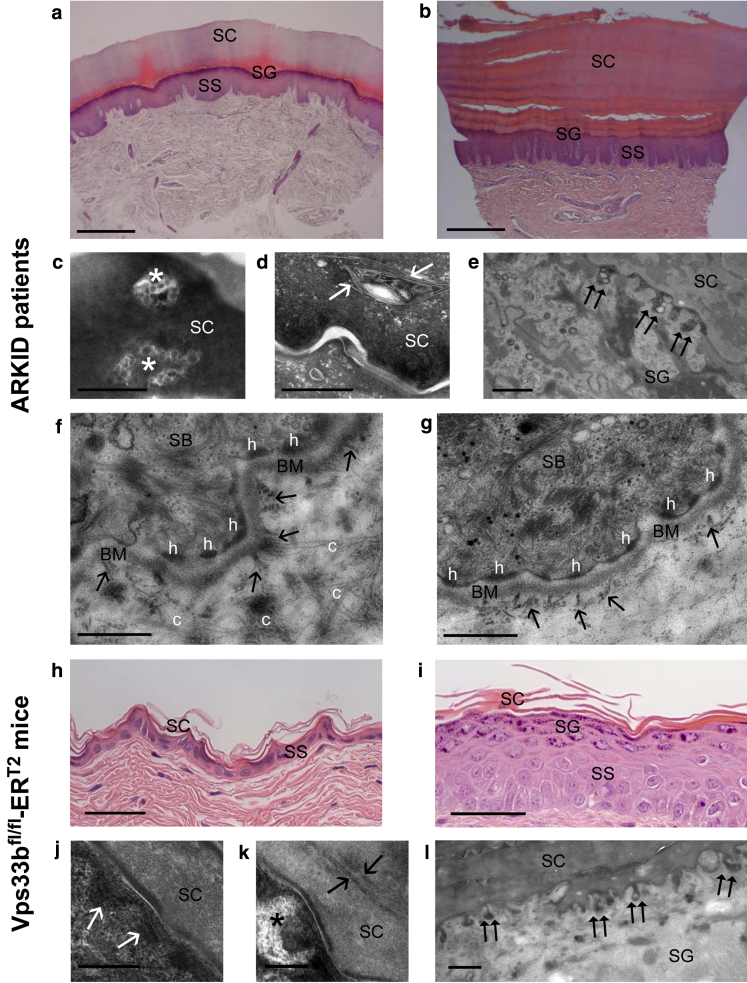
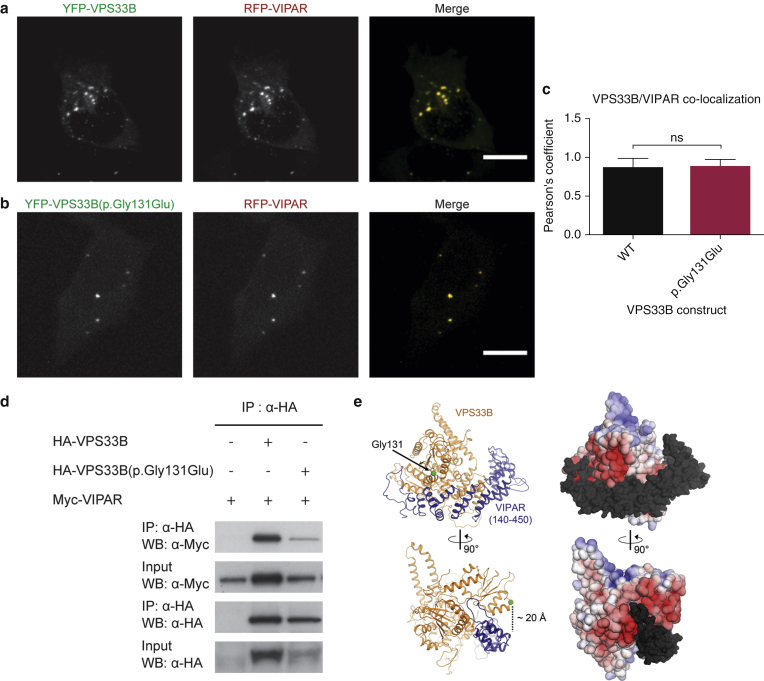
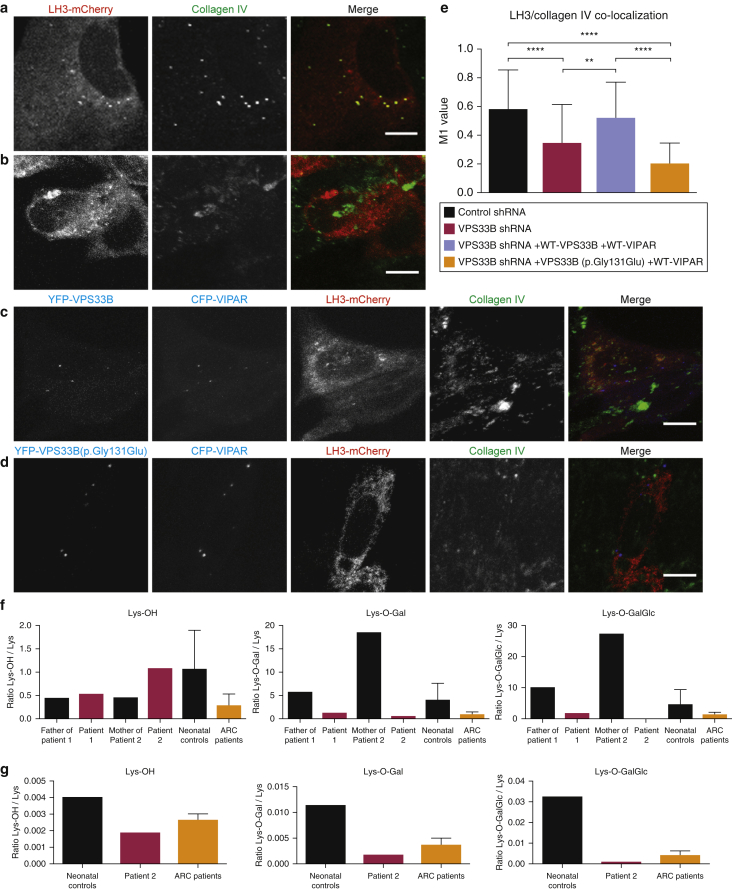
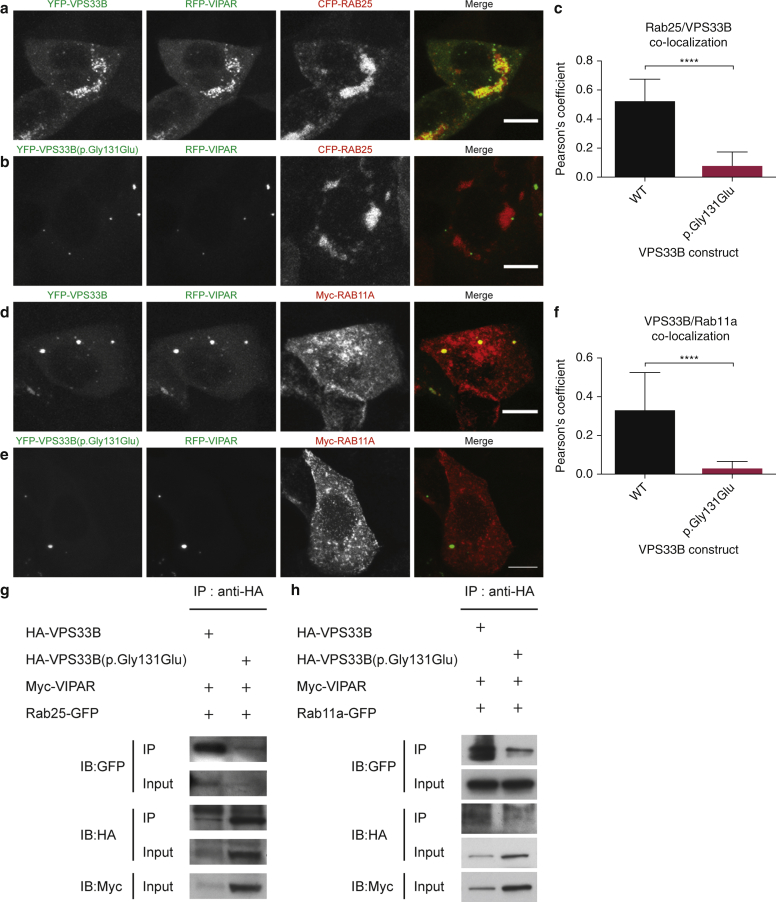
In this paper, we report three patients with severe palmoplantar keratoderma associated with ichthyosis and sensorineural deafness. Biallelic mutations were found in VPS33B, encoding VPS33B, a Sec1/Munc18 family protein that interacts with Rab11a and Rab25 proteins and is involved in trafficking of the collagen-modifying enzyme LH3. Two patients were homozygous for the missense variant p.Gly131Glu, whereas one patient was compound heterozygous for p.Gly131Glu and the splice site mutation c.240-1G>C, previously reported in patients with arthrogryposis renal dysfunction and cholestasis syndrome. We demonstrated the pathogenicity of variant p.Gly131Glu by assessing the interactions of the mutant VPS33B construct and its ability to traffic LH3. Compared with wild-type VPS33B, the p.Gly131Glu mutant VPS33B had reduced coimmunoprecipitation and colocalization with Rab11a and Rab25 and did not rescue LH3 trafficking. Confirming the cell-based experiments, we found deficient LH3-specific collagen lysine modifications in patients' urine and skin fibroblasts. Additionally, the epidermal ultrastructure of the p.Gly131Glu patients mirrored defects in tamoxifen-inducible VPS33B-deficient Vps33b-ER mice. Both patients and murine models revealed an impaired epidermal structure, ascribed to aberrant secretion of lamellar bodies, which are essential for epidermal barrier formation. Our results demonstrate that p.Gly131Glu mutant VPS33B causes an autosomal recessive keratoderma-ichthyosis-deafness syndrome.
在本文中,我们报告了3例患有严重掌跖角化病并伴有鱼鳞病和感音神经性耳聋的患者。在编码VPS33B的VPS33B基因中发现了双等位基因突变,VPS33B是一种Sec1/Munc18家族蛋白,可与Rab11a和Rab25蛋白相互作用,并参与胶原修饰酶LH3的运输。2例患者为错义变体p.Gly131Glu纯合子,而1例患者为p.Gly131Glu与剪接位点突变c.240-1G>C的复合杂合子,该突变先前在关节挛缩、肾功能不全和胆汁淤积综合征患者中报道过。我们通过评估突变型VPS33B构建体的相互作用及其运输LH3的能力,证实了变体p.Gly131Glu的致病性。与野生型VPS33B相比,p.Gly131Glu突变型VPS33B与Rab11a和Rab25的共免疫沉淀和共定位减少,并且不能挽救LH3的运输。在细胞实验得到证实后,我们在患者尿液和皮肤成纤维细胞中发现了LH3特异性胶原赖氨酸修饰缺陷。此外,p.Gly131Glu患者的表皮超微结构反映了他莫昔芬诱导的VPS33B缺陷型Vps33b-ER小鼠的缺陷。患者和小鼠模型均显示表皮结构受损,这归因于对表皮屏障形成至关重要的板层小体分泌异常。我们的结果表明,p.Gly131Glu突变型VPS33B导致常染色体隐性角化病-鱼鳞病-耳聋综合征。