Basha Gutierrez Josep, Nakai Kenta
Department of Computational Biology and Medical Sciences, Graduate School of Frontier Sciences, The University of Tokyo, 277-8561, Chiba, Japan.
Human Genome Center, The Institute of Medical Science, The University of Tokyo, 4-6-1 Shirokane-dai, Minato-ku, 108-8639, Tokyo, Japan.
BMC Bioinformatics. 2016 Dec 22;17(Suppl 19):502. doi: 10.1186/s12859-016-1364-3.
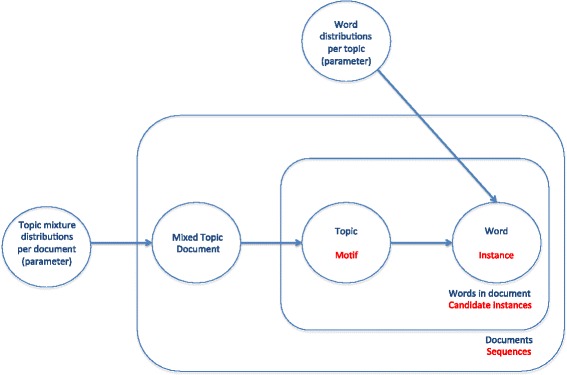
Topic models are statistical algorithms which try to discover the structure of a set of documents according to the abstract topics contained in them. Here we try to apply this approach to the discovery of the structure of the transcription factor binding sites (TFBS) contained in a set of biological sequences, which is a fundamental problem in molecular biology research for the understanding of transcriptional regulation. Here we present two methods that make use of topic models for motif finding. First, we developed an algorithm in which first a set of biological sequences are treated as text documents, and the k-mers contained in them as words, to then build a correlated topic model (CTM) and iteratively reduce its perplexity. We also used the perplexity measurement of CTMs to improve our previous algorithm based on a genetic algorithm and several statistical coefficients.
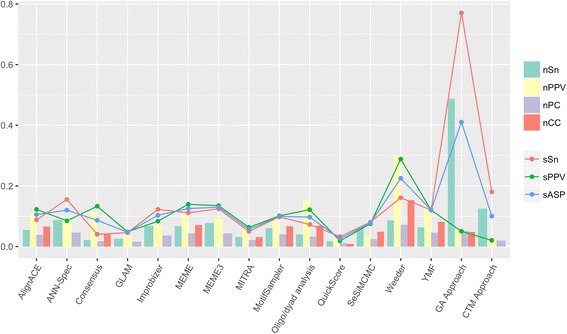
The algorithms were tested with 56 data sets from four different species and compared to 14 other methods by the use of several coefficients both at nucleotide and site level. The results of our first approach showed a performance comparable to the other methods studied, especially at site level and in sensitivity scores, in which it scored better than any of the 14 existing tools. In the case of our previous algorithm, the new approach with the addition of the perplexity measurement clearly outperformed all of the other methods in sensitivity, both at nucleotide and site level, and in overall performance at site level.
The statistics obtained show that the performance of a motif finding method based on the use of a CTM is satisfying enough to conclude that the application of topic models is a valid method for developing motif finding algorithms. Moreover, the addition of topic models to a previously developed method dramatically increased its performance, suggesting that this combined algorithm can be a useful tool to successfully predict motifs in different kinds of sets of DNA sequences.
主题模型是一种统计算法,旨在根据文档中包含的抽象主题来发现一组文档的结构。在此,我们尝试将这种方法应用于发现一组生物序列中所含转录因子结合位点(TFBS)的结构,这是分子生物学研究中理解转录调控的一个基本问题。我们在此介绍两种利用主题模型进行基序查找的方法。首先,我们开发了一种算法,先将一组生物序列视为文本文档,将其中包含的k-mer视为单词,然后构建相关主题模型(CTM)并迭代降低其困惑度。我们还利用CTM的困惑度测量来改进我们之前基于遗传算法和几个统计系数的算法。
使用来自四个不同物种的56个数据集对算法进行了测试,并通过核苷酸和位点水平的几个系数与其他14种方法进行了比较。我们第一种方法的结果显示出与其他研究方法相当的性能,尤其是在位点水平和灵敏度得分方面,其得分优于14种现有工具中的任何一种。对于我们之前的算法,添加了困惑度测量的新方法在核苷酸和位点水平的灵敏度以及位点水平的整体性能方面明显优于所有其他方法。
获得的统计数据表明,基于使用CTM的基序查找方法的性能足够令人满意,足以得出主题模型的应用是开发基序查找算法的有效方法这一结论。此外,将主题模型添加到先前开发的方法中显著提高了其性能,表明这种组合算法可以成为成功预测不同类型DNA序列集中基序的有用工具。