Department of Pharmacometrics and Pharmacokinetics, Meiji Pharmaceutical University, 2-522-1 Noshio, Kiyose, Tokyo, 204-8588, Japan.
Drugs R D. 2017 Sep;17(3):475-480. doi: 10.1007/s40268-017-0193-9.
Human α1-acid glycoprotein has genetic variants, the F1, S, and A variants, which can be separated isoelectrophoretically. These variants show differences in their affinity of binding to several drugs. In this study, we investigated the factors determining drug binding to these α1-acid glycoprotein genetic variants using disopyramide, warfarin, and tamsulosin as marker compounds.
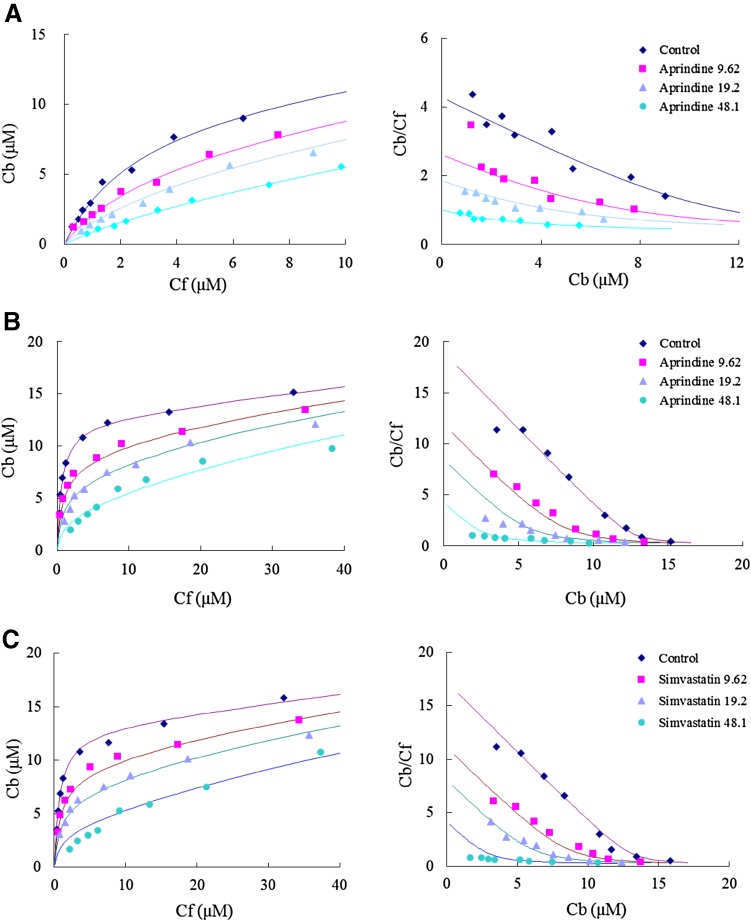
Binding of the marker drugs to human α1-acid glycoprotein was determined by ultra-filtration in the presence or absence of various other drugs. For screening of the α1-acid glycoprotein variants to which the marker drugs became bound, the effects of various other drugs on their binding were studied. The binding data were analyzed using a competitive inhibition model and the relationship between the estimated dissociation constants and physicochemical properties, such as log P, was also analyzed.
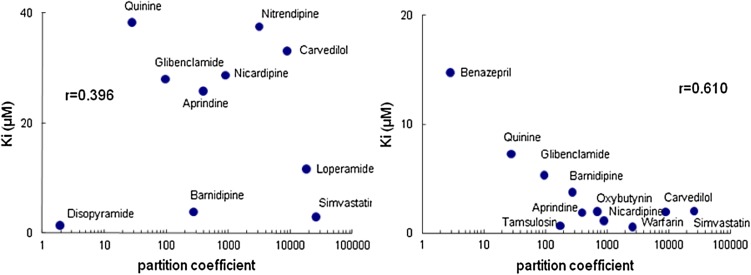
The binding of tamsulosin was significantly decreased by aprindine, carvedilol, erythromycin, thioridazine, and warfarin, but not by disopyramide. The dissociation constants of drugs bound to F1/S variants were significantly correlated with their lipophilicity, but those for the A variant were not.
We were able to develop a simple screening method for determining individual α1-acid glycoprotein variants to which drugs would bind. The binding of drugs to F1/S variants may be determined mainly by drug lipophilicity.
人α1-酸性糖蛋白存在遗传变异体 F1、S 和 A 型,可通过等电聚焦电泳分离。这些变异体在与几种药物的结合亲和力上存在差异。本研究使用地尔硫卓、华法林和坦索罗辛作为标记化合物,探讨了决定这些α1-酸性糖蛋白遗传变异体与药物结合的因素。
通过超滤法在存在或不存在各种其他药物的情况下,测定标记药物与人类α1-酸性糖蛋白的结合。为筛选与标记药物结合的α1-酸性糖蛋白变异体,研究了各种其他药物对其结合的影响。使用竞争抑制模型分析结合数据,并分析估计的解离常数与物理化学性质(如 log P)之间的关系。
阿普林定、卡维地洛、红霉素、硫利达嗪和华法林显著降低了坦索罗辛的结合,而地尔硫卓则没有。与 F1/S 变异体结合的药物的解离常数与它们的亲脂性显著相关,但与 A 变异体的解离常数无关。
我们能够开发出一种简单的筛选方法,用于确定药物将结合的个体α1-酸性糖蛋白变异体。药物与 F1/S 变异体的结合可能主要取决于药物的亲脂性。