Kawarai Toshitaka, Morigaki Ryoma, Kaji Ryuji, Goto Satoshi
Department of Clinical Neuroscience, Institute of Biomedical Sciences, Graduate School of Medical Sciences, Tokushima University, Tokushima 770-8503, Japan.
Parkinson's Disease and Dystonia Research Center, Tokushima University Hospital, Tokushima 770-8503, Japan.
Brain Sci. 2017 Jun 26;7(7):72. doi: 10.3390/brainsci7070072.
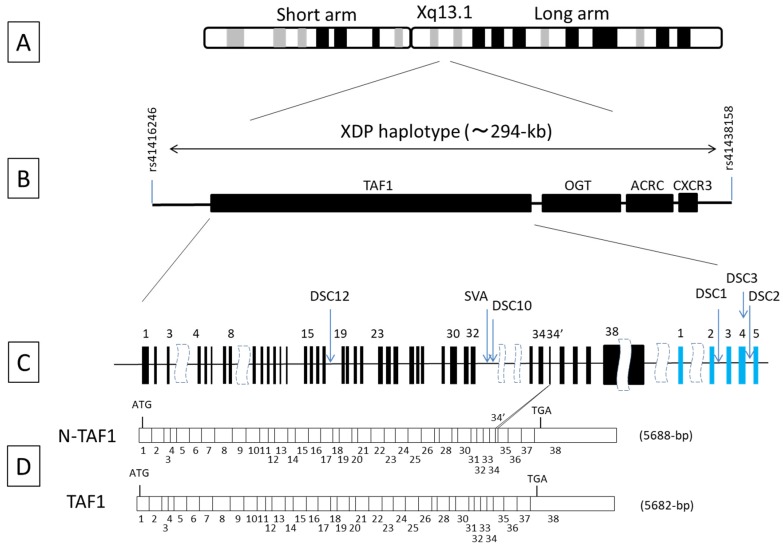
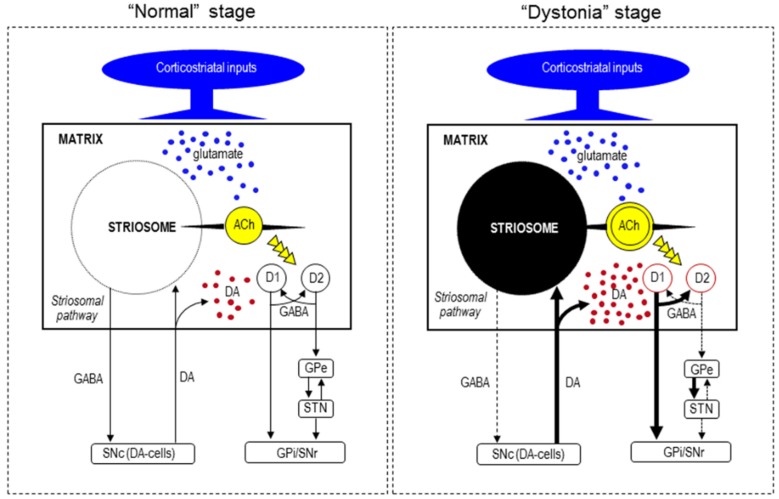
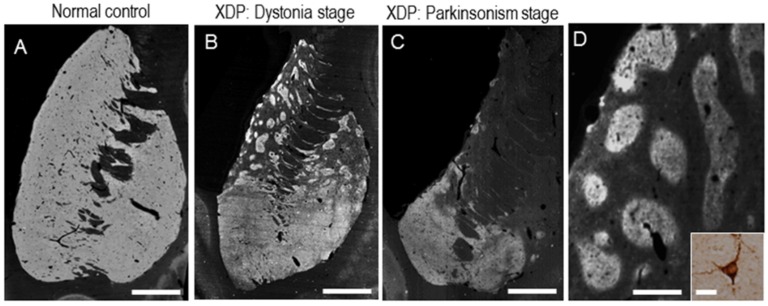
X-linked dystonia-parkinsonism (XDP; OMIM314250), also referred to as DYT3 dystonia or "Lubag" disease, was first described as an endemic disease in the Philippine island of Panay. XDP is an adult-onset movement disorder characterized by progressive and severe dystonia followed by overt parkinsonism in the later years of life. Among the primary monogenic dystonias, XDP has been identified as a transcriptional dysregulation syndrome with impaired expression of the (TATA box-binding protein associated factor 1) gene, which is a critical component of the cellular transcription machinery. The major neuropathology of XDP is progressive neuronal loss in the neostriatum (i.e., the caudate nucleus and putamen). XDP may be used as a human disease model to elucidate the pathomechanisms by which striatal neurodegeneration leads to dystonia symptoms. In this article, we introduce recent advances in the understanding of the interplay between pathophysiology and genetics in XDP.
X连锁肌张力障碍-帕金森综合征(XDP;OMIM314250),也被称为DYT3肌张力障碍或“Lubag”病,最初被描述为菲律宾班乃岛的一种地方病。XDP是一种成人起病的运动障碍,其特征是进行性严重肌张力障碍,随后在生命后期出现明显的帕金森综合征。在原发性单基因肌张力障碍中,XDP已被确定为一种转录失调综合征,其(TATA盒结合蛋白相关因子1)基因表达受损,该基因是细胞转录机制的关键组成部分。XDP的主要神经病理学表现为新纹状体(即尾状核和壳核)中进行性神经元丢失。XDP可作为一种人类疾病模型,以阐明纹状体神经变性导致肌张力障碍症状的病理机制。在本文中,我们介绍了对XDP病理生理学与遗传学之间相互作用的最新认识进展。