Department of Translational Neuroscience, Osaka City University Graduate School of Medicine, 1-4-3 Asahimachi, Abeno-ku, Osaka, 545-8585, Japan.
Core Research for Evolutional Science and Technology, Japan Science and Technology Agency, Kawaguchi, Japan.
Acta Neuropathol Commun. 2017 Jul 31;5(1):59. doi: 10.1186/s40478-017-0461-5.
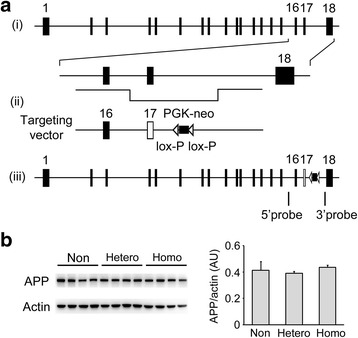
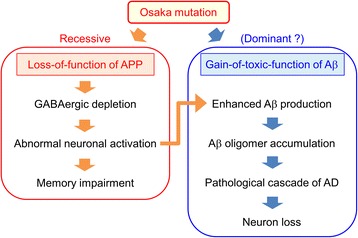
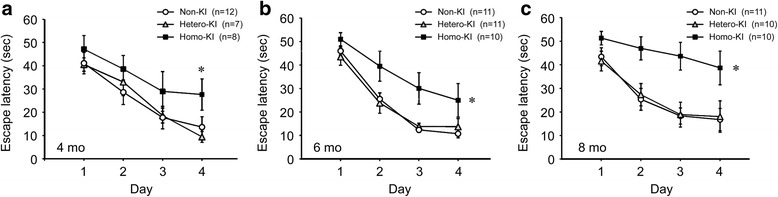
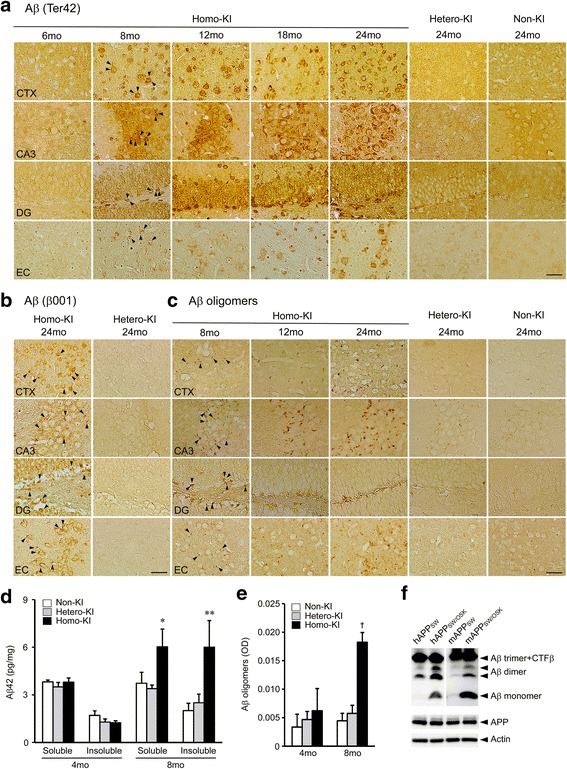
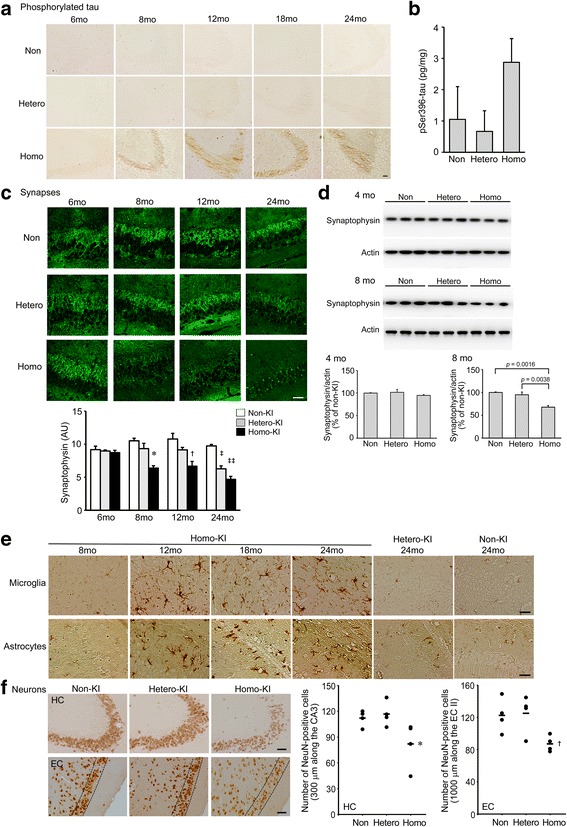
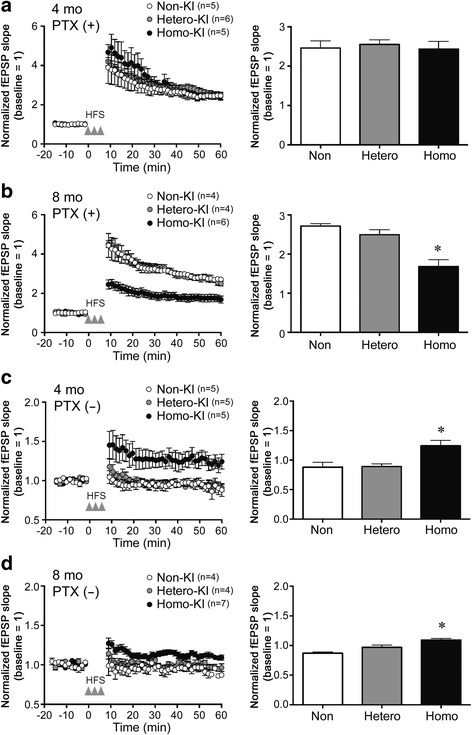
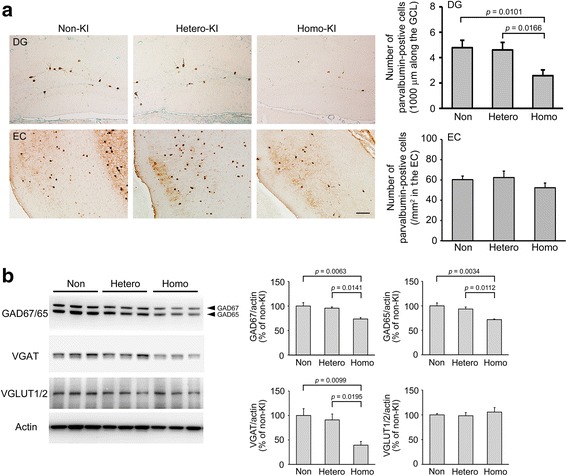
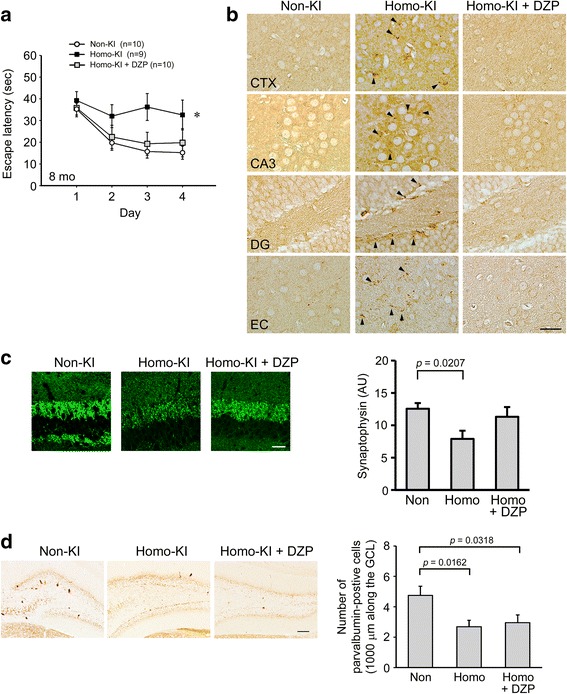
The E693Δ (Osaka) mutation in APP is linked to familial Alzheimer's disease. While this mutation accelerates amyloid β (Aβ) oligomerization, only patient homozygotes suffer from dementia, implying that this mutation is recessive and causes loss-of-function of amyloid precursor protein (APP). To investigate the recessive trait, we generated a new mouse model by knocking-in the Osaka mutation into endogenous mouse APP. The produced homozygous, heterozygous, and non-knockin littermates were compared for memory, neuropathology, and synaptic plasticity. Homozygotes showed memory impairment at 4 months, whereas heterozygotes did not, even at 8 months. Immunohistochemical and biochemical analyses revealed that only homozygotes displayed intraneuronal accumulation of Aβ oligomers at 8 months, followed by abnormal tau phosphorylation, synapse loss, glial activation, and neuron loss. These pathologies were not observed at younger ages, suggesting that a certain mechanism other than Aβ accumulation underlies the memory disturbance at 4 months. For the electrophysiology studies at 4 months, high-frequency stimulation evoked long-term potentiation in all mice in the presence of picrotoxin, but in the absence of picrotoxin, such potentiation was observed only in homozygotes, suggesting their GABAergic deficit. In support of this, the levels of GABA-related proteins and the number of dentate GABAergic interneurons were decreased in 4-month-old homozygotes. Since APP has been shown to play a role in dentate GABAergic synapse formation, the observed GABAergic depletion is likely associated with an impairment of the APP function presumably caused by the Osaka mutation. Oral administration of diazepam to homozygotes from 6 months improved memory at 8 months, and furthermore, prevented Aβ oligomer accumulation, indicating that GABAergic deficiency is a cause of memory impairment and also a driving force of Aβ accumulation. Our findings suggest that the Osaka mutation causes loss of APP function, leading to GABAergic depletion and memory disorder when wild-type APP is absent, providing a mechanism of the recessive heredity.
E693Δ(大阪)突变 APP 与家族性阿尔茨海默病有关。虽然这种突变会加速淀粉样 β(Aβ)寡聚体的形成,但只有患者纯合子才会患痴呆症,这表明这种突变是隐性的,会导致淀粉样前体蛋白(APP)功能丧失。为了研究隐性特征,我们通过将大阪突变敲入内源性小鼠 APP 生成了一种新的小鼠模型。比较了产生的纯合子、杂合子和非敲入同窝仔鼠的记忆、神经病理学和突触可塑性。纯合子在 4 个月时表现出记忆障碍,而杂合子即使在 8 个月时也没有。免疫组织化学和生化分析表明,只有纯合子在 8 个月时显示出 Aβ寡聚物在神经元内的积累,随后出现异常的 tau 磷酸化、突触丢失、胶质细胞激活和神经元丢失。这些病理变化在较年轻时没有观察到,这表明在 4 个月时记忆障碍的原因不是 Aβ的积累。对于 4 个月时的电生理学研究,在皮卡毒素存在的情况下,高频刺激可诱发所有小鼠的长时程增强,但在没有皮卡毒素的情况下,只有纯合子观察到这种增强,表明它们存在 GABA 能缺陷。支持这一观点的是,4 个月大的纯合子中 GABA 相关蛋白的水平和齿状 GABA 能中间神经元的数量减少。由于 APP 已被证明在齿状 GABA 能突触形成中发挥作用,因此观察到的 GABA 能耗竭可能与大阪突变引起的 APP 功能障碍有关。从 6 个月开始,给纯合子口服地西泮可改善 8 个月时的记忆,并且还可以防止 Aβ寡聚体的积累,这表明 GABA 能缺陷是记忆障碍的原因,也是 Aβ积累的驱动力。我们的研究结果表明,大阪突变导致 APP 功能丧失,当野生型 APP 不存在时,导致 GABA 能耗竭和记忆障碍,提供了隐性遗传的机制。