Genome Institute of Singapore, Singapore, 138672, Singapore.
Institute of Molecular and Cell Biology, Singapore, 138673, Singapore.
BMC Genomics. 2017 Oct 27;18(1):829. doi: 10.1186/s12864-017-4217-1.
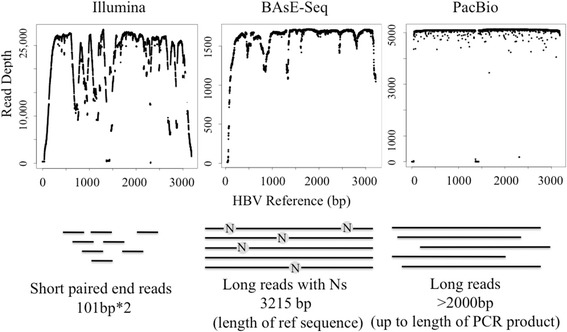
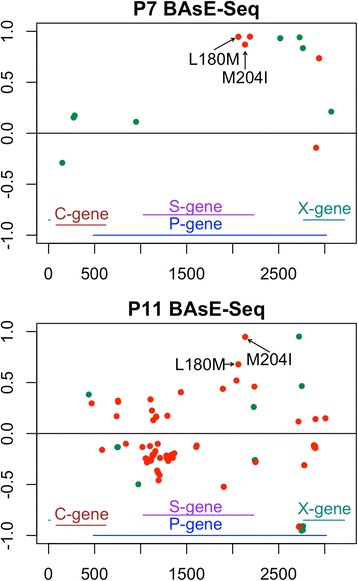
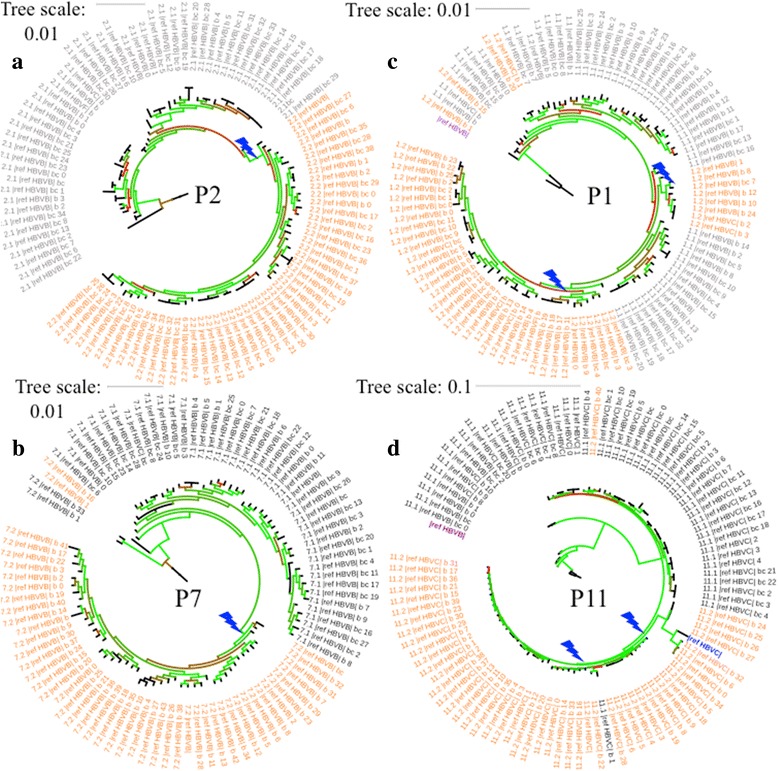
Viral populations are complex, dynamic, and fast evolving. The evolution of groups of closely related viruses in a competitive environment is termed quasispecies. To fully understand the role that quasispecies play in viral evolution, characterizing the trajectories of viral genotypes in an evolving population is the key. In particular, long-range haplotype information for thousands of individual viruses is critical; yet generating this information is non-trivial. Popular deep sequencing methods generate relatively short reads that do not preserve linkage information, while third generation sequencing methods have higher error rates that make detection of low frequency mutations a bioinformatics challenge. Here we applied BAsE-Seq, an Illumina-based single-virion sequencing technology, to eight samples from four chronic hepatitis B (CHB) patients - once before antiviral treatment and once after viral rebound due to resistance.
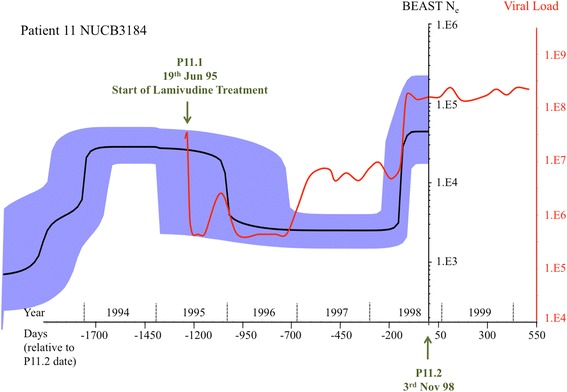
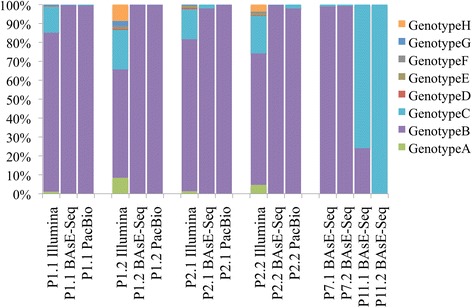
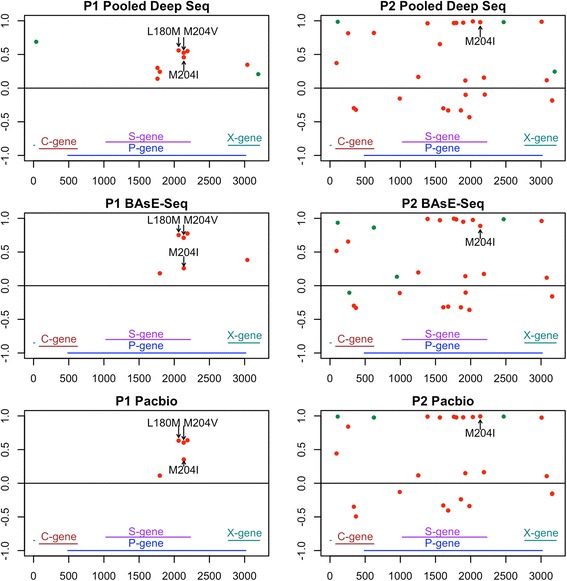
With single-virion sequencing, we obtained 248-8796 single-virion sequences per sample, which allowed us to find evidence for both hard and soft selective sweeps. We were able to reconstruct population demographic history that was independently verified by clinically collected data. We further verified four of the samples independently through PacBio SMRT and Illumina Pooled deep sequencing.
Overall, we showed that single-virion sequencing yields insight into viral evolution and population dynamics in an efficient and high throughput manner. We believe that single-virion sequencing is widely applicable to the study of viral evolution in the context of drug resistance and host adaptation, allows differentiation between soft or hard selective sweeps, and may be useful in the reconstruction of intra-host viral population demographic history.
病毒群体复杂、动态且快速进化。在竞争环境中,密切相关的病毒群体的进化被称为准种。为了充分了解准种在病毒进化中的作用,对进化群体中病毒基因型的轨迹进行特征描述是关键。特别是,需要大量个体病毒的长程单倍型信息;然而,生成此信息并非易事。流行的深度测序方法生成的相对较短的读取序列无法保留连接信息,而第三代测序方法的错误率更高,使得检测低频突变成为一个生物信息学挑战。在这里,我们应用了基于 Illumina 的单病毒测序技术 BAsE-Seq,对来自四名慢性乙型肝炎 (CHB) 患者的八个样本进行了研究——一次是在抗病毒治疗前,一次是在因耐药性而发生病毒反弹后。
通过单病毒测序,我们每个样本获得了 248-8796 条单病毒序列,这使我们能够找到硬选择和软选择的证据。我们能够重建人口动态历史,该历史通过临床收集的数据进行了独立验证。我们还通过 PacBio SMRT 和 Illumina Pooled 深度测序独立验证了其中四个样本。
总体而言,我们表明单病毒测序以高效和高通量的方式深入了解病毒进化和群体动态。我们认为单病毒测序广泛适用于耐药和宿主适应背景下的病毒进化研究,能够区分硬选择或软选择,并且可能有助于重建宿主内病毒群体的人口动态历史。