Chen Chao-Jin, Liu De-Zhao, Yao Wei-Feng, Gu Yu, Huang Fei, Hei Zi-Qing, Li Xiang
Department of Anesthesiology, The Third Affiliated Hospital of Sun Yat-sen University, Guangzhou, People's Republic of China.
J Pain Res. 2017 Nov 14;10:2665-2674. doi: 10.2147/JPR.S143431. eCollection 2017.
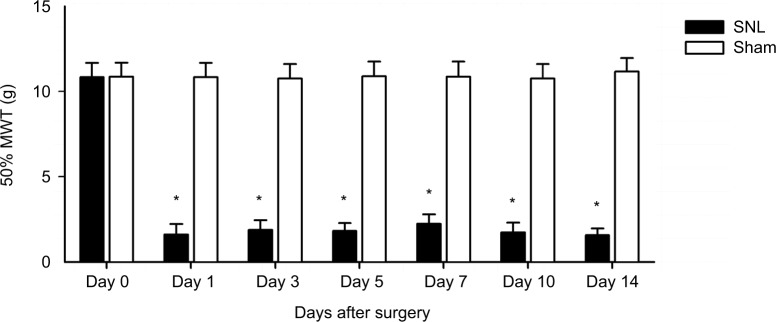
Neuropathic pain is a complex chronic condition occurring post-nervous system damage. The transcriptional reprogramming of injured dorsal root ganglia (DRGs) drives neuropathic pain. However, few comparative analyses using high-throughput platforms have investigated uninjured DRG in neuropathic pain, and potential interactions among differentially expressed genes (DEGs) and pathways were not taken into consideration. The aim of this study was to identify changes in genes and pathways associated with neuropathic pain in uninjured L4 DRG after L5 spinal nerve ligation (SNL) by using bioinformatic analysis.
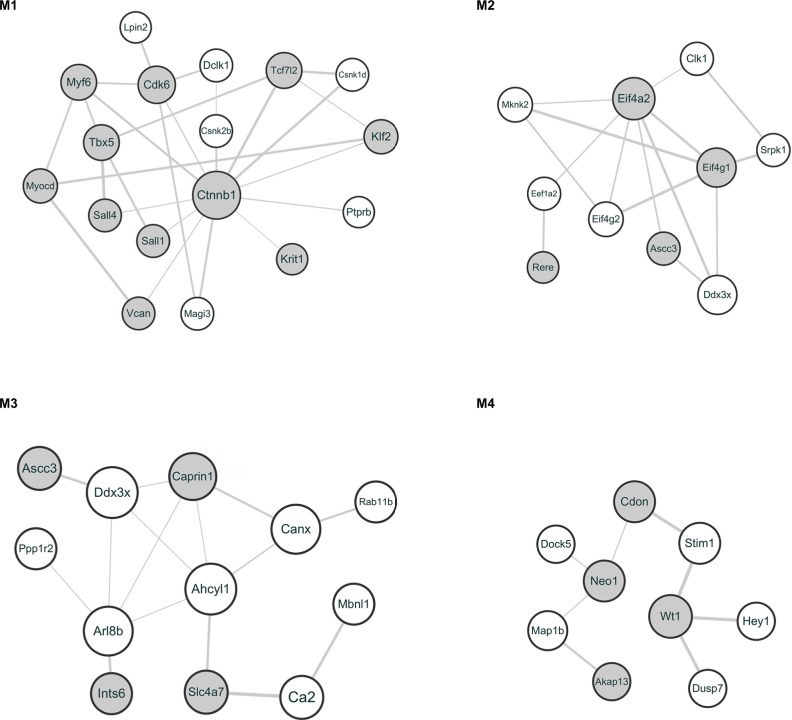
The microarray profile GSE24982 was downloaded from the Gene Expression Omnibus database to identify DEGs between DRGs in SNL and sham rats. The prioritization for these DEGs was performed using the Toppgene database followed by gene ontology and pathway enrichment analyses. The relationships among DEGs from the protein interactive perspective were analyzed using protein-protein interaction (PPI) network and module analysis. Real-time polymerase chain reaction (PCR) and Western blotting were used to confirm the expression of DEGs in the rodent neuropathic pain model.
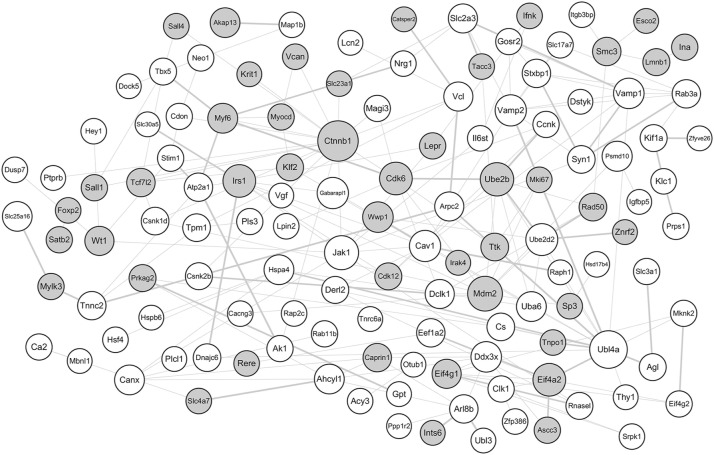
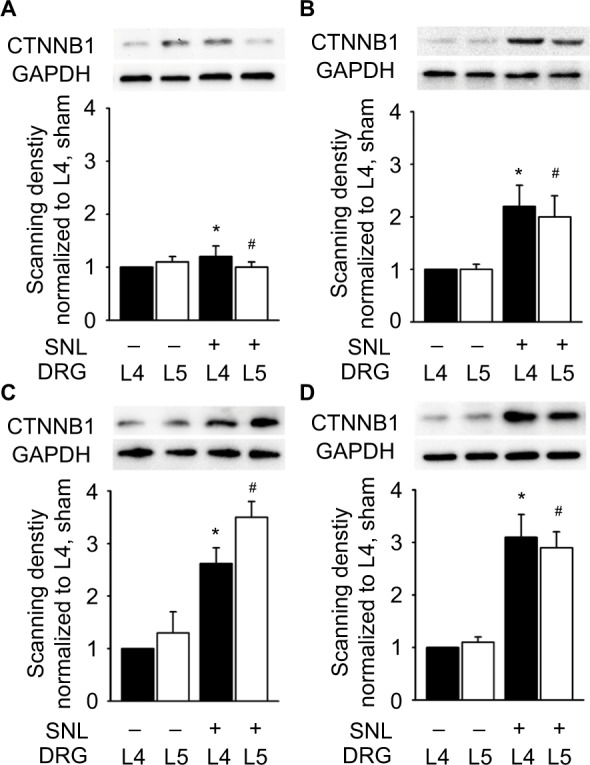
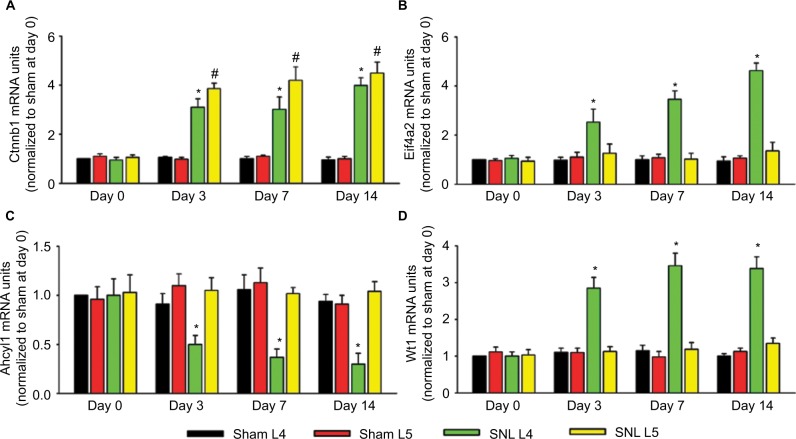
A total of 206 DEGs that might play a role in neuropathic pain were identified in L4 DRG, of which 75 were upregulated and 131 were downregulated. The upregulated DEGs were enriched in biological processes related to transcription regulation and molecular functions such as DNA binding, cell cycle, and the FoxO signaling pathway. Ctnnb1 protein had the highest connectivity degrees in the PPI network. The in vivo studies also validated that mRNA and protein levels of Ctnnb1 were upregulated in both L4 and L5 DRGs.
This study provides insight into the functional gene sets and pathways associated with neuropathic pain in L4 uninjured DRG after L5 SNL, which might promote our understanding of the molecular mechanisms underlying the development of neuropathic pain.
神经性疼痛是神经系统损伤后出现的一种复杂的慢性病症。受损背根神经节(DRG)的转录重编程引发神经性疼痛。然而,很少有使用高通量平台的比较分析研究神经性疼痛中未受损的DRG,并且未考虑差异表达基因(DEG)和通路之间的潜在相互作用。本研究的目的是通过生物信息学分析确定L5脊神经结扎(SNL)后未受损的L4 DRG中与神经性疼痛相关的基因和通路的变化。
从基因表达综合数据库下载微阵列图谱GSE24982,以鉴定SNL大鼠和假手术大鼠DRG之间的DEG。使用Toppgene数据库对这些DEG进行优先级排序,随后进行基因本体和通路富集分析。从蛋白质相互作用的角度分析DEG之间的关系,使用蛋白质-蛋白质相互作用(PPI)网络和模块分析。使用实时聚合酶链反应(PCR)和蛋白质印迹法确认啮齿动物神经性疼痛模型中DEG的表达。
在L4 DRG中鉴定出总共206个可能在神经性疼痛中起作用的DEG,其中75个上调,131个下调。上调的DEG富集于与转录调控相关的生物学过程以及诸如DNA结合、细胞周期和FoxO信号通路等分子功能。Ctnnb1蛋白在PPI网络中具有最高的连接度。体内研究还证实,L4和L5 DRG中Ctnnb1的mRNA和蛋白水平均上调。
本研究深入了解了L5 SNL后L4未受损DRG中与神经性疼痛相关的功能基因集和通路,这可能促进我们对神经性疼痛发生发展分子机制的理解。