Ungar Rachel A, Giri Neelam, Pao Maryland, Khincha Payal P, Zhou Weiyin, Alter Blanche P, Savage Sharon A
Clinical Genetics Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute (NCI), National Institutes of Health, Rockville, Maryland.
National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland.
Am J Med Genet A. 2018 Jun;176(6):1432-1437. doi: 10.1002/ajmg.a.38706. Epub 2018 Apr 25.
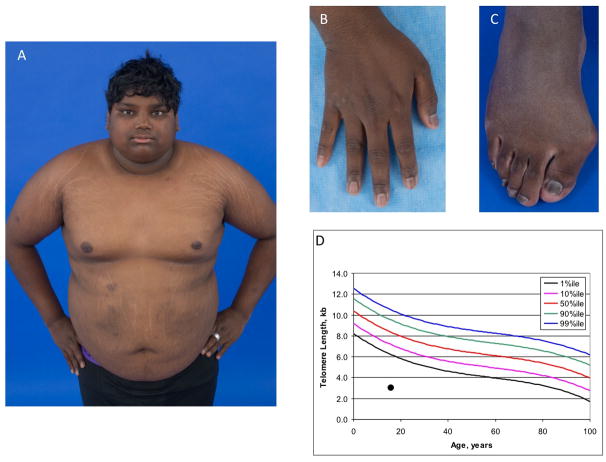
Dyskeratosis congenita (DC) is an inherited bone marrow failure syndrome caused by germline mutations in telomere biology genes. Patients have extremely short telomeres for their age and a complex phenotype including oral leukoplakia, abnormal skin pigmentation, and dysplastic nails in addition to bone marrow failure, pulmonary fibrosis, stenosis of the esophagus, lacrimal ducts and urethra, developmental anomalies, and high risk of cancer. We evaluated a patient with features of DC, mood dysregulation, diabetes, and lack of pubertal development. Family history was not available but genome-wide genotyping was consistent with consanguinity. Whole exome sequencing identified 82 variants of interest in 80 genes based on the following criteria: homozygous, <0.1% minor allele frequency in public and in-house databases, nonsynonymous, and predicted deleterious by multiple in silico prediction programs. Six genes were identified likely contributory to the clinical presentation. The cause of DC is likely due to homozygous splice site variants in regulator of telomere elongation helicase 1, a known DC and telomere biology gene. A homozygous, missense variant in tryptophan hydroxylase 1 may be clinically important as this gene encodes the rate limiting step in serotonin biosynthesis, a biologic pathway connected with mood disorders. Four additional genes (SCN4A, LRP4, GDAP1L1, and SPTBN5) had rare, missense homozygous variants that we speculate may contribute to portions of the clinical phenotype. This case illustrates the value of conducting detailed clinical and genomic evaluations on rare patients in order to identify new areas of research into the functional consequences of rare variants and their contribution to human disease.
先天性角化不良(DC)是一种遗传性骨髓衰竭综合征,由端粒生物学基因的种系突变引起。患者在其年龄阶段具有极短的端粒,并且具有复杂的表型,除了骨髓衰竭、肺纤维化、食管、泪管和尿道狭窄、发育异常以及癌症高风险外,还包括口腔白斑、皮肤色素沉着异常和甲发育异常。我们评估了一名具有DC特征、情绪调节障碍、糖尿病和青春期发育缺失的患者。家族史不详,但全基因组基因分型显示与近亲结婚相符。全外显子测序基于以下标准在80个基因中鉴定出82个感兴趣的变异:纯合、在公共和内部数据库中的次要等位基因频率<0.1%、非同义,并且被多个计算机预测程序预测为有害。确定了六个可能导致临床表现的基因。DC的病因可能是端粒延伸解旋酶1(一种已知的DC和端粒生物学基因)中的纯合剪接位点变异。色氨酸羟化酶1中的一个纯合错义变异可能具有临床重要性,因为该基因编码血清素生物合成中的限速步骤,血清素生物合成是一条与情绪障碍相关的生物学途径。另外四个基因(SCN4A、LRP4、GDAP1L1和SPTBN5)具有罕见的纯合错义变异,我们推测这些变异可能导致部分临床表型。该病例说明了对罕见患者进行详细临床和基因组评估的价值,以便确定关于罕见变异的功能后果及其对人类疾病的贡献的新研究领域。