MRC Human Genetics Unit, MRC Institute of Genetics and Molecular Medicine, The University of Edinburgh, Western General Hospital, Crewe Road, Edinburgh, EH4 2XU, UK.
Hum Genet. 2019 Sep;138(8-9):881-898. doi: 10.1007/s00439-018-1934-8. Epub 2018 Sep 22.
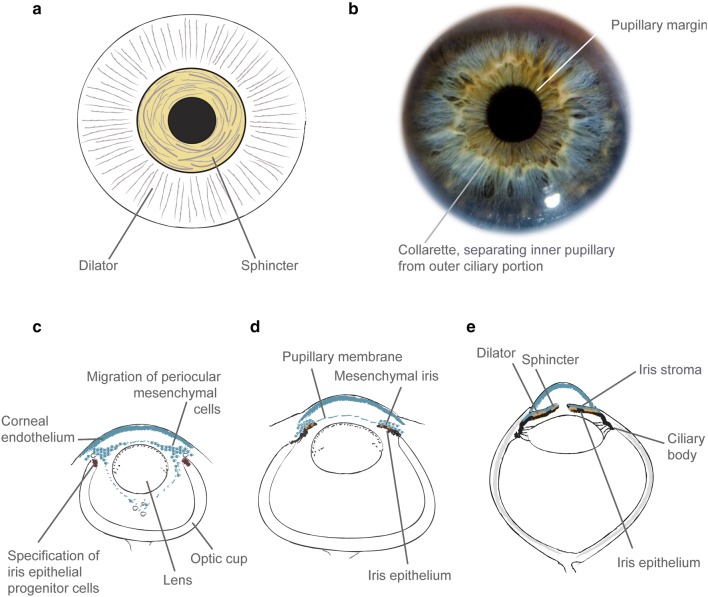
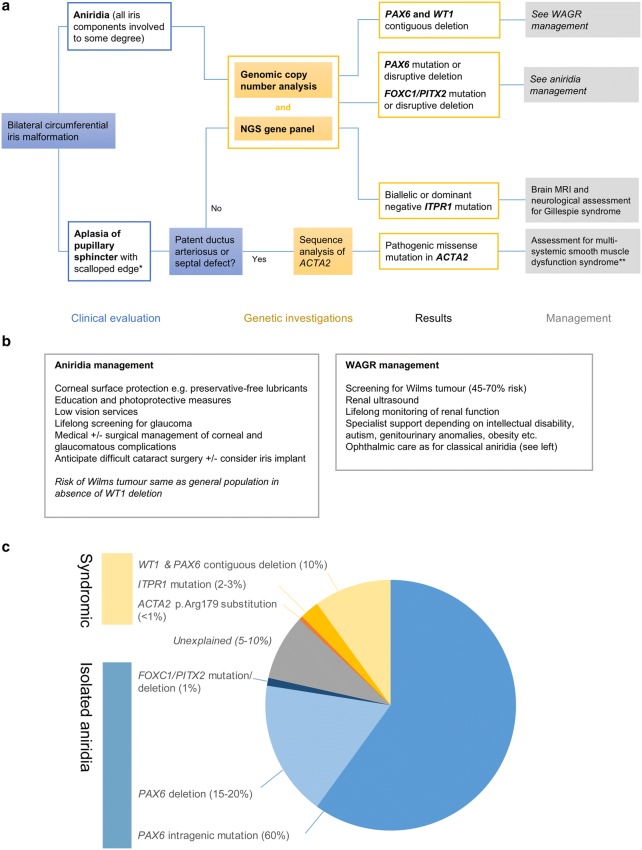
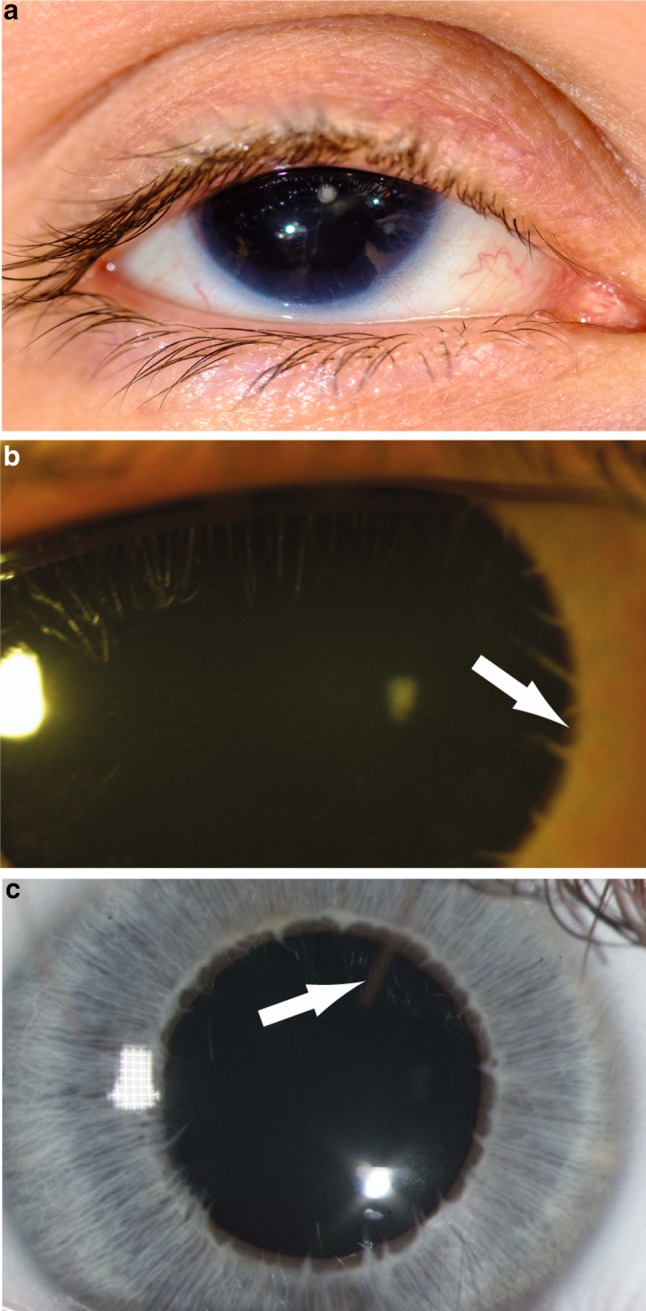
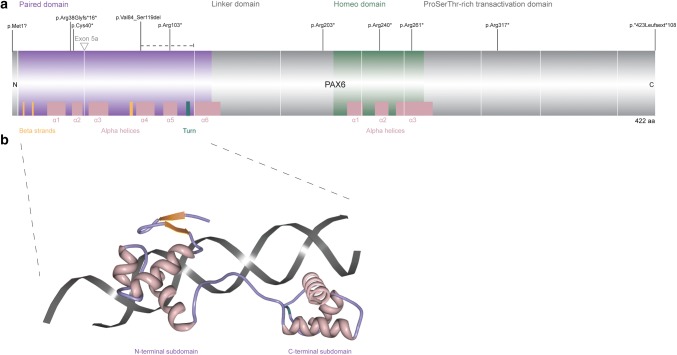
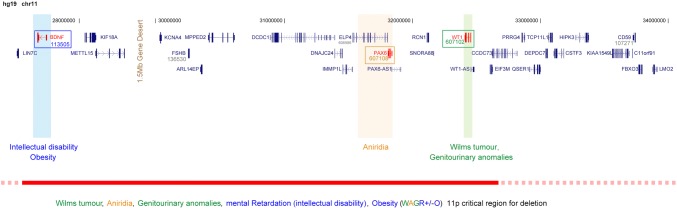
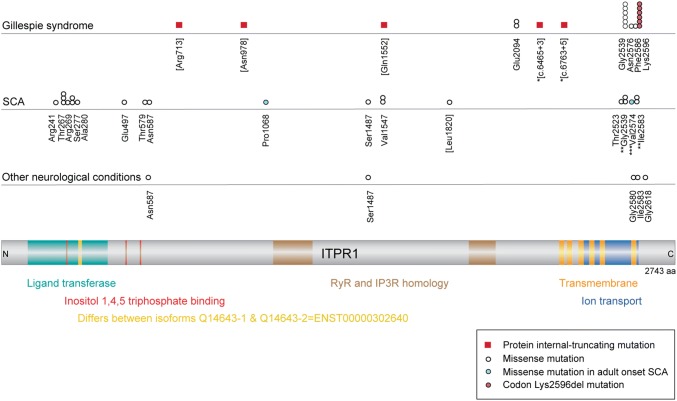
Absence of part or all of the iris, aniridia, is a feature of several genetically distinct conditions. This review focuses on iris development and then the clinical features and molecular genetics of these iris malformations. Classical aniridia, a panocular eye malformation including foveal hypoplasia, is the archetypal phenotype associated with heterozygous PAX6 loss-of-function mutations. Since this was identified in 1991, many genetic mechanisms of PAX6 inactivation have been elucidated, the commonest alleles being intragenic mutations causing premature stop codons, followed by those causing C-terminal extensions. Rarely, aniridia cases are associated with FOXC1, PITX2 and/or their regulatory regions. Aniridia can also occur as a component of many severe global eye malformations. Gillespie syndrome-a triad of partial aniridia, non-progressive cerebellar ataxia and intellectual disability-is phenotypically and genotypically distinct from classical aniridia. The causative gene has recently been identified as ITPR1. The same characteristic Gillespie syndrome-like iris, with aplasia of the pupillary sphincter and a scalloped margin, is seen in ACTA2-related multisystemic smooth muscle dysfunction syndrome. WAGR syndrome (Wilms tumour, aniridia, genitourinary anomalies and mental retardation/intellectual disability), is caused by contiguous deletion of PAX6 and WT1 on chromosome 11p. Deletions encompassing BDNF have been causally implicated in the obesity and intellectual disability associated with the condition. Lastly, we outline a genetic investigation strategy for aniridia in light of recent developments, suggesting an approach based principally on chromosomal array and gene panel testing. This strategy aims to test all known aniridia loci-including the rarer, life-limiting causes-whilst remaining simple and practical.
虹膜部分或全部缺失,即无虹膜,是几种具有不同遗传特征的疾病的特征。本篇综述聚焦于虹膜的发育,然后介绍这些虹膜畸形的临床表现和分子遗传学。经典的无虹膜是一种眼球多器官畸形,包括中心凹发育不良,是与 PAX6 功能丧失杂合突变相关的典型表型。自 1991 年发现以来,许多 PAX6 失活的遗传机制已经阐明,最常见的等位基因是导致提前终止密码子的基因内突变,其次是导致 C 末端延伸的突变。很少情况下,无虹膜病例与 FOXC1、PITX2 和/或它们的调节区相关。无虹膜也可能是许多严重的眼球整体畸形的一部分。Gillespie 综合征——部分无虹膜、进行性小脑共济失调和智力障碍三联征——在表型和基因型上与经典无虹膜不同。最近,其致病基因已被确定为 ITPR1。同样具有 Gillespie 综合征特征的虹膜,伴有瞳孔括约肌发育不全和扇形边缘,也见于 ACTA2 相关的多系统平滑肌功能障碍综合征。WAGR 综合征(Wilms 瘤、无虹膜、泌尿生殖系统异常和智力低下/智力残疾)是由染色体 11p 上 PAX6 和 WT1 的连续缺失引起的。包含 BDNF 的缺失被认为与该疾病相关的肥胖和智力残疾有关。最后,我们根据最新进展概述了无虹膜的遗传研究策略,建议基于染色体微阵列和基因面板测试的方法。该策略旨在测试所有已知的无虹膜基因座,包括较罕见的、危及生命的病因,同时保持简单实用。