Hashikami Kentarou, Asahina Makoto, Nozu Kandai, Iijima Kazumoto, Nagata Michio, Takeyama Michiyasu
Pharmaceutical Research Division, Takeda Pharmaceutical Company Limited, 2-26-1, Muraoka-Higashi, Fujisawa, Kanagawa, 251-8555, Japan.
Department of Pediatrics, Kobe University Graduate School of Medicine, Kobe, Hyogo 651-0017, Japan.
Biochem Biophys Rep. 2018 Dec 12;17:81-86. doi: 10.1016/j.bbrep.2018.12.003. eCollection 2019 Mar.
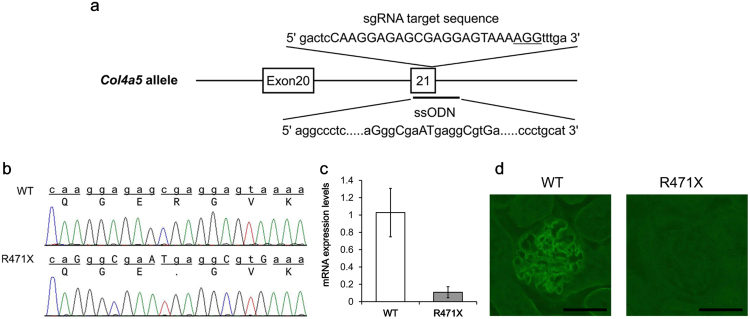
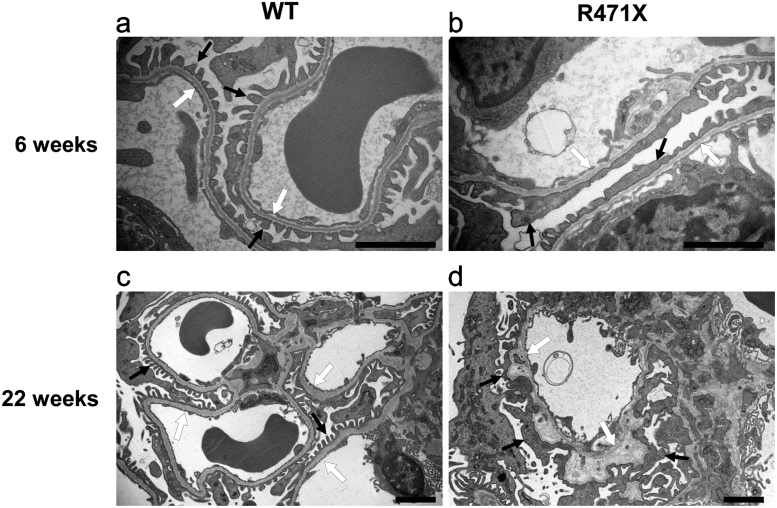
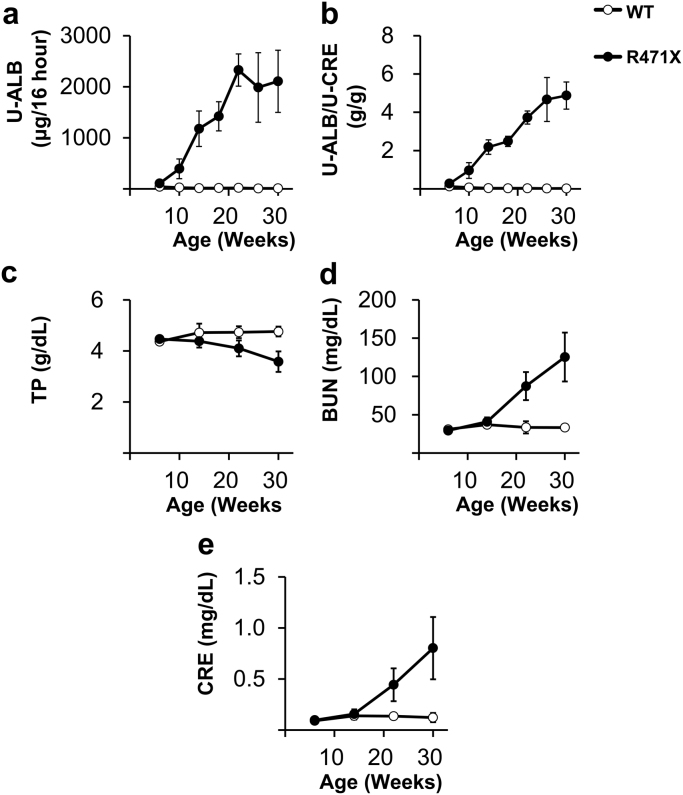
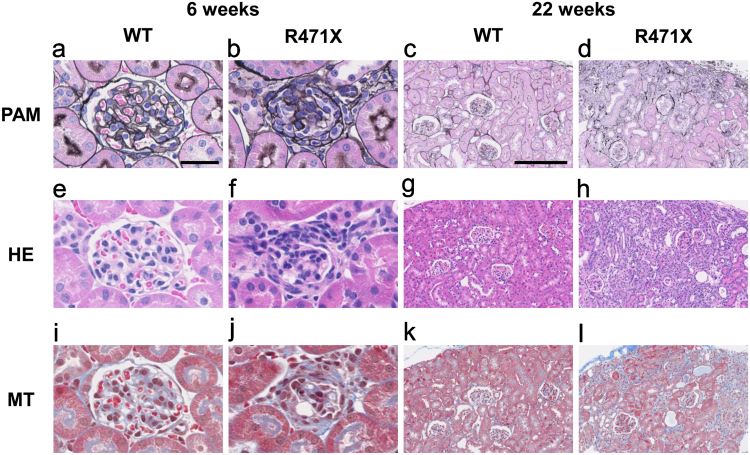
Alport syndrome (AS) is an inherited disorder characterized by glomerular basement membrane (GBM) abnormality and development of chronic kidney disease at an early age. The cause of AS is a genetic mutation in type IV collagen, and more than 80% of patients have X-linked AS (XLAS) with mutation in . Although the causal gene has been identified, mechanisms of progression have not been elucidated, and no effective treatment has been developed. In this study, we generated a mutant mouse harboring a nonsense mutation (R471X) obtained from a patient with XLAS using clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated system. mRNA and protein expressions were not observed in the kidneys of hemizygous R471X male mice. R471X mice showed proteinuria and hematuria. Pathology revealed progression of glomerulosclerosis and interstitial fibrosis by age. Electron microscopy identified irregular thickening in GBM accompanied by irregular lamination. These observations were consistent with the clinical and pathological features of patients with AS and other established models. In addition, our mice models develop end-stage renal disease at the median age of 28 weeks, much later compared to previous models much more consistent with clinical course of human XLAS. Our models have advantages for future experiments in regard with treatment for human XLAS.
阿尔波特综合征(AS)是一种遗传性疾病,其特征为肾小球基底膜(GBM)异常以及早年出现慢性肾脏病。AS的病因是IV型胶原基因突变,超过80%的患者患有X连锁AS(XLAS),其 发生突变。尽管致病基因已被确定,但疾病进展机制尚未阐明,也未开发出有效的治疗方法。在本研究中,我们使用成簇规律间隔短回文重复序列(CRISPR)/CRISPR相关系统,构建了一只携带从一名XLAS患者获得的无义突变(R471X)的突变小鼠。在半合子R471X雄性小鼠的肾脏中未观察到 mRNA和蛋白表达。R471X小鼠出现蛋白尿和血尿。病理学检查显示,随着年龄增长,肾小球硬化和间质纤维化逐渐进展。电子显微镜检查发现GBM不规则增厚并伴有不规则分层。这些观察结果与AS患者及其他已建立模型的临床和病理特征一致。此外,我们的小鼠模型在28周龄的中位年龄时发展为终末期肾病,与先前模型相比要晚得多,这与人类XLAS的临床病程更为一致。我们的模型在未来针对人类XLAS治疗的实验方面具有优势。