Department of Medicine, University of Cambridge, Addenbrooke's Hospital, Hills Road, Cambridge, UK.
MRC Biostatistics Unit, Institute of Public Health, University Forvie Site, Robinson Way, Cambridge, UK.
BMC Genomics. 2019 Jan 23;20(1):77. doi: 10.1186/s12864-018-5314-5.
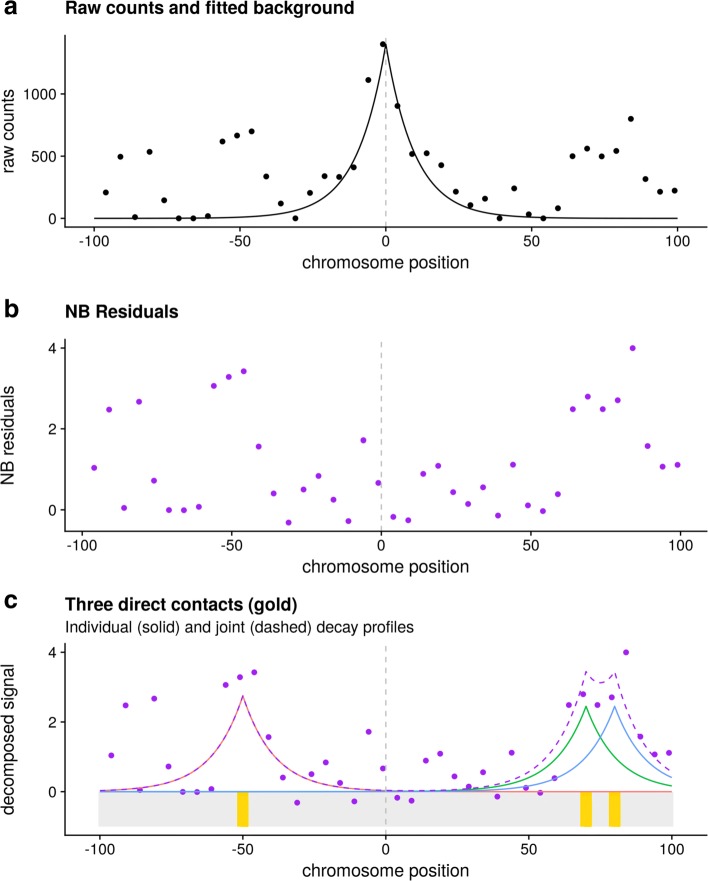
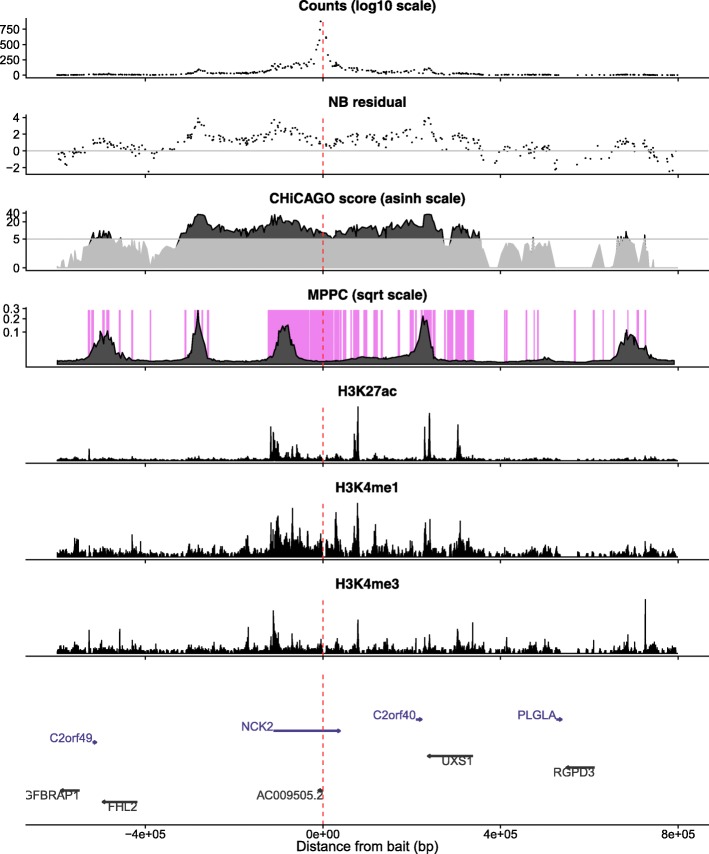
Hi-C and capture Hi-C (CHi-C) are used to map physical contacts between chromatin regions in cell nuclei using high-throughput sequencing. Analysis typically proceeds considering the evidence for contacts between each possible pair of fragments independent from other pairs. This can produce long runs of fragments which appear to all make contact with the same baited fragment of interest.
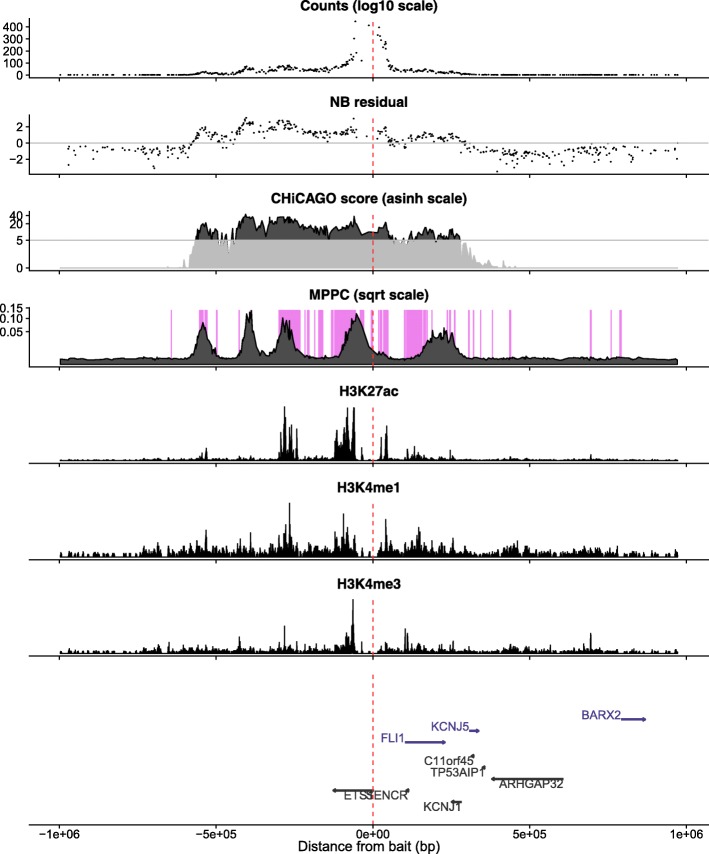
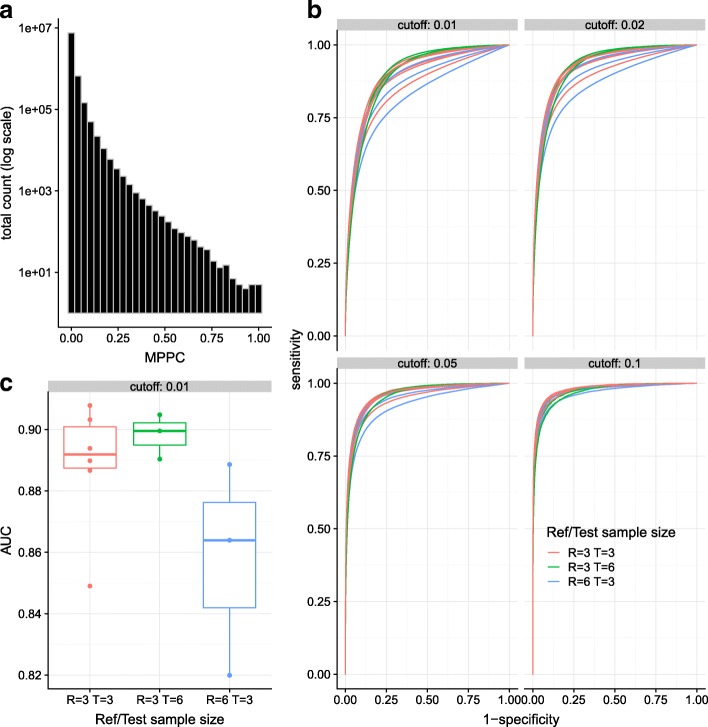
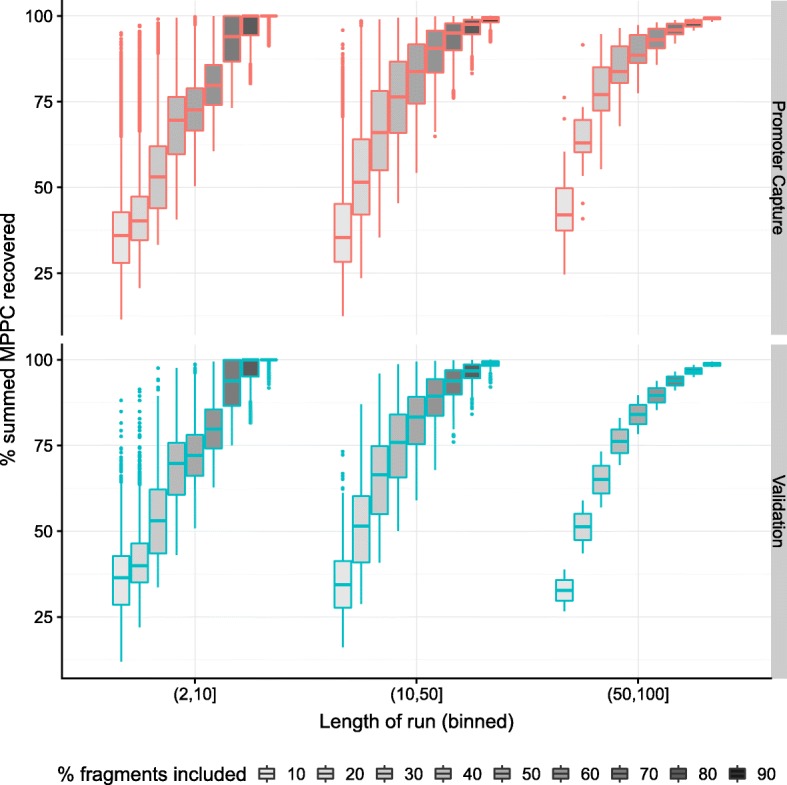
We hypothesised that these long runs could result from a smaller subset of direct contacts and propose a new method, based on a Bayesian sparse variable selection approach, which attempts to fine map these direct contacts. Our model is conceptually novel, exploiting the spatial pattern of counts in CHi-C data. Although we use only the CHi-C count data in fitting the model, we show that the fragments prioritised display biological properties that would be expected of true contacts: for bait fragments corresponding to gene promoters, we identify contact fragments with active chromatin and contacts that correspond to edges found in previously defined enhancer-target networks; conversely, for intergenic bait fragments, we identify contact fragments corresponding to promoters for genes expressed in that cell type. We show that long runs of apparently co-contacting fragments can typically be explained using a subset of direct contacts consisting of <10% of the number in the full run, suggesting that greater resolution can be extracted from existing datasets.
Our results appear largely complementary to those from a per-fragment analytical approach, suggesting that they provide an additional level of interpretation that may be used to increase resolution for mapping direct contacts in CHi-C experiments.
Hi-C 和捕获 Hi-C(CHi-C)用于使用高通量测序绘制细胞核中染色质区域之间的物理接触图谱。分析通常考虑每对片段之间接触证据的独立于其他对的证据进行。这可能会产生看起来都与相同诱饵片段接触的长片段。
我们假设这些长片段可能来自于更小的直接接触子集,并提出了一种新方法,该方法基于贝叶斯稀疏变量选择方法,试图对这些直接接触进行精细映射。我们的模型在概念上是新颖的,利用了 CHi-C 数据中计数的空间模式。虽然我们仅在拟合模型时使用 CHi-C 计数数据,但我们表明,优先排序的片段显示出预期的真实接触的生物学特性:对于对应于基因启动子的诱饵片段,我们识别出与活性染色质接触的片段,以及与先前定义的增强子-靶标网络中发现的边缘对应的接触;相反,对于基因间诱饵片段,我们识别出与该细胞类型中表达的基因的启动子对应的接触片段。我们表明,明显共同接触的片段的长片段通常可以使用直接接触的子集来解释,该子集由完整运行中片段数的<10%组成,这表明可以从现有数据集中提取更高的分辨率。
我们的结果在很大程度上与基于片段的分析方法的结果互补,表明它们提供了额外的解释水平,可用于提高 CHi-C 实验中直接接触的分辨率。