Rana Rabia Mukhtar, Rampogu Shailima, Zeb Amir, Son Minky, Park Chanin, Lee Gihwan, Yoon Sanghwa, Baek Ayoung, Parameswaran Sarvanan, Park Seok Ju, Lee Keun Woo
Division of Life Sciences, Division of Applied Life Science (BK21 Plus), Research Institute of NaturalScience (RINS), Gyeongsang National University (GNU), 501 Jinju-daero, Jinju 52828, Korea.
Department of Internal Medicine, College of Medicine, Busan Paik Hospital, Inje University,Busan 47392, Korea.
J Clin Med. 2019 Feb 11;8(2):233. doi: 10.3390/jcm8020233.
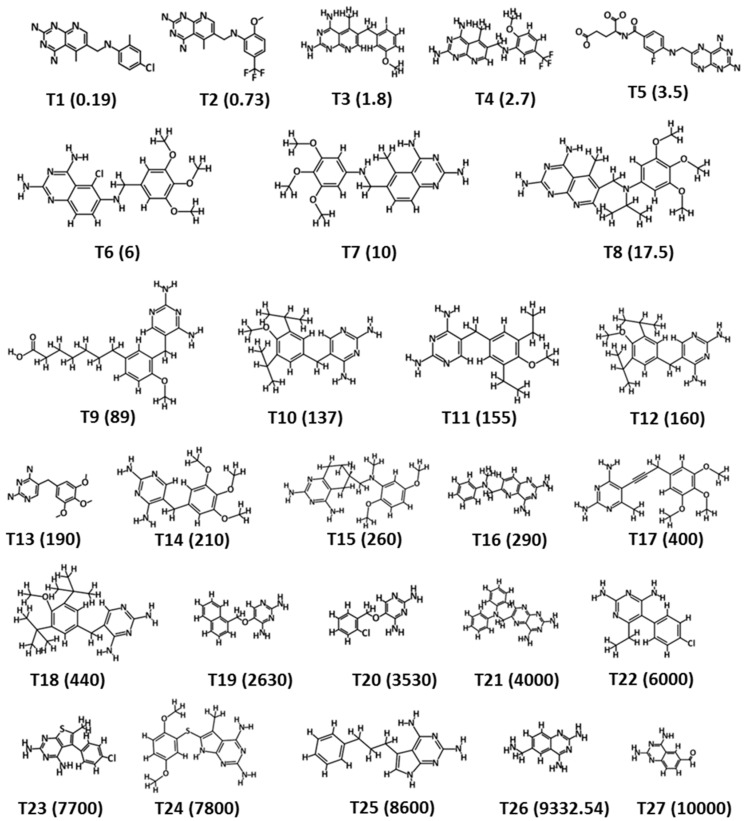
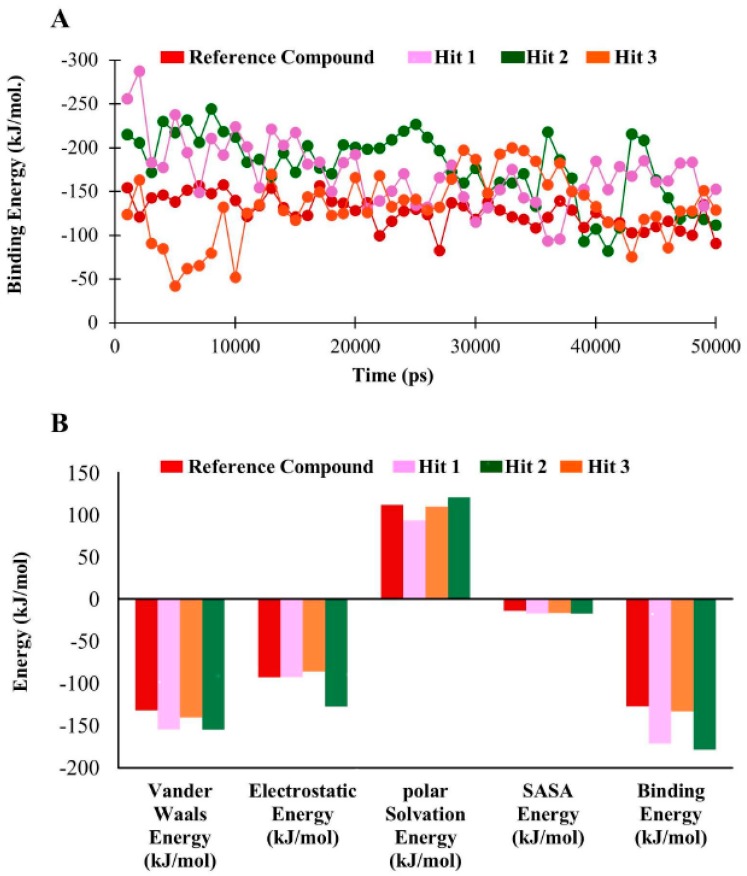
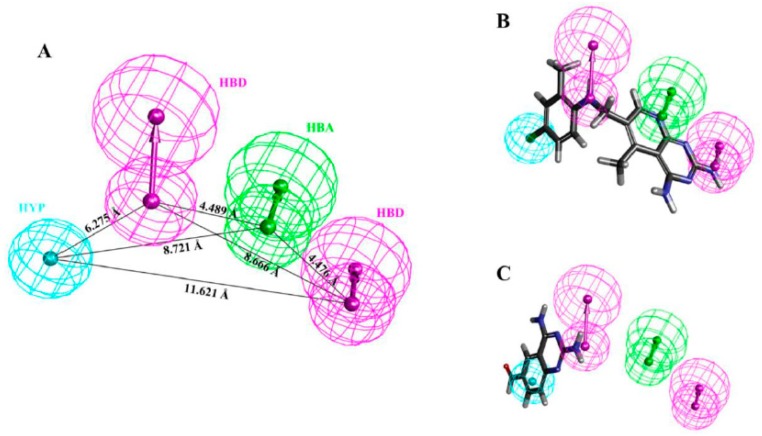
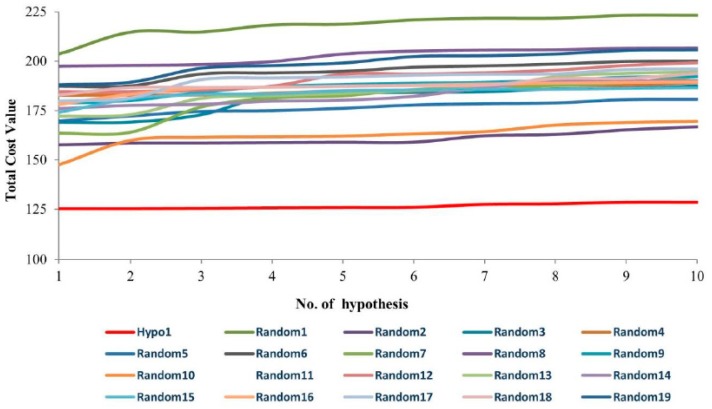
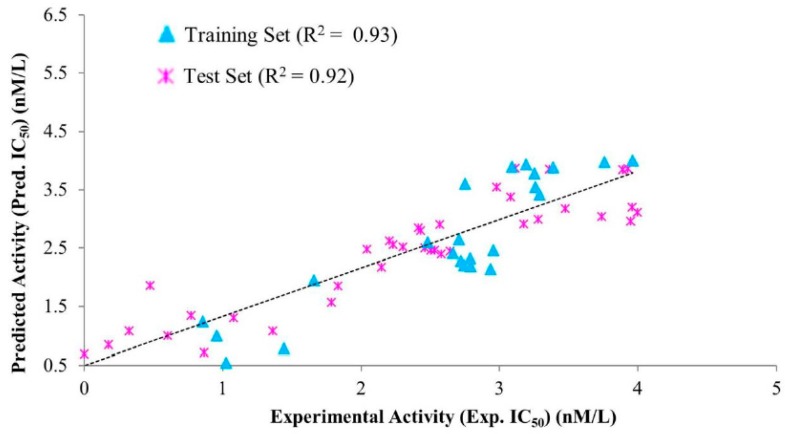
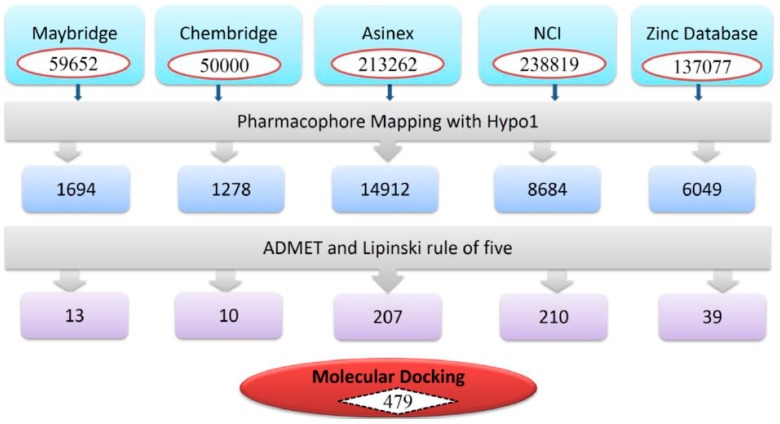
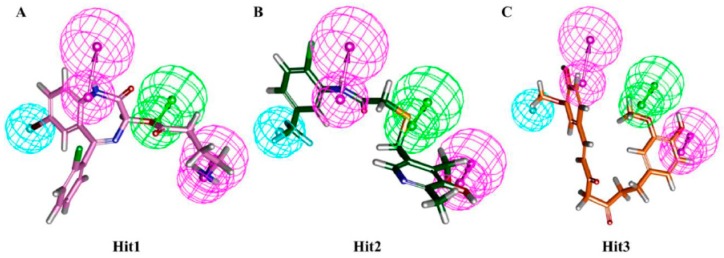
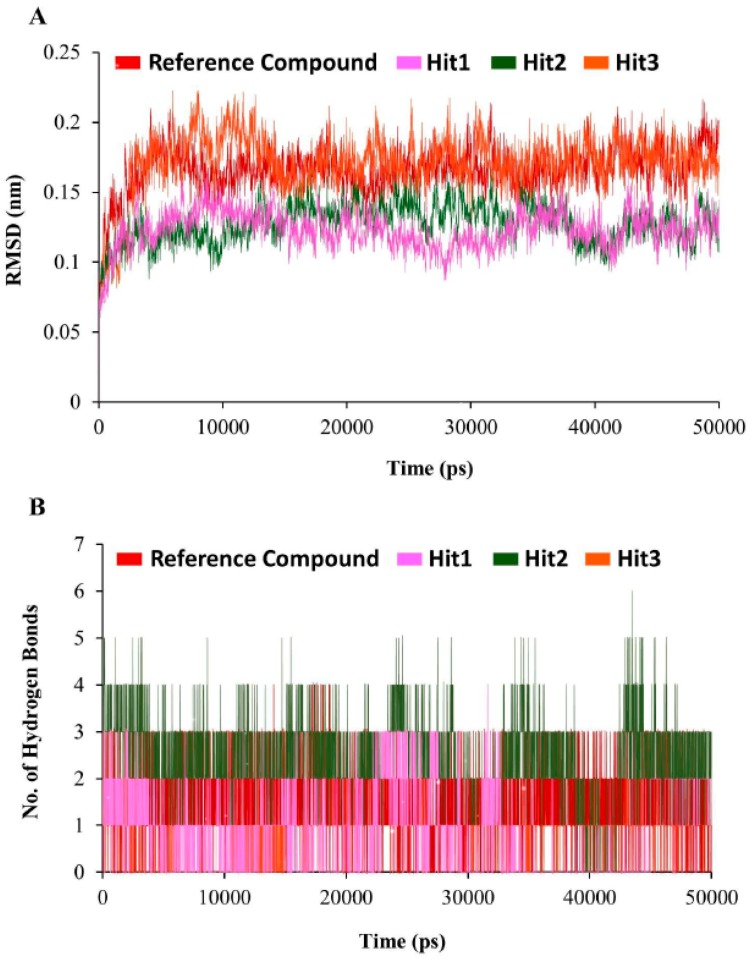
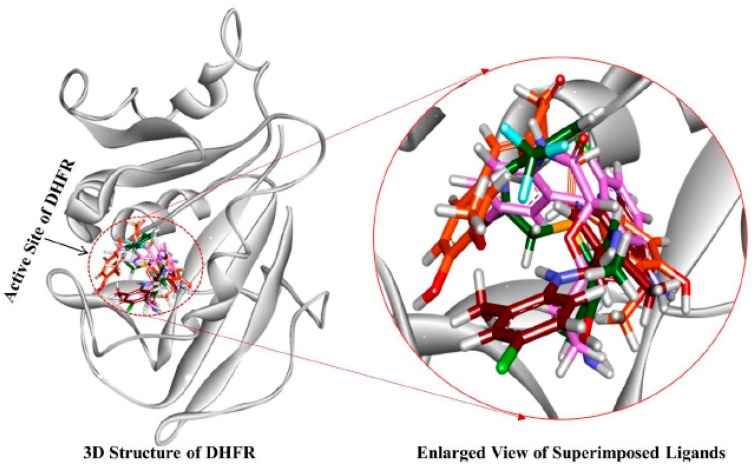
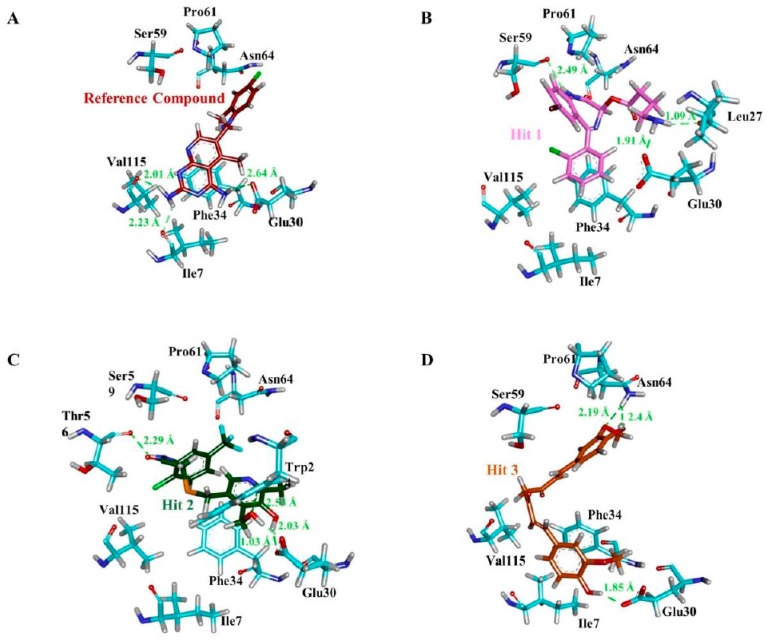
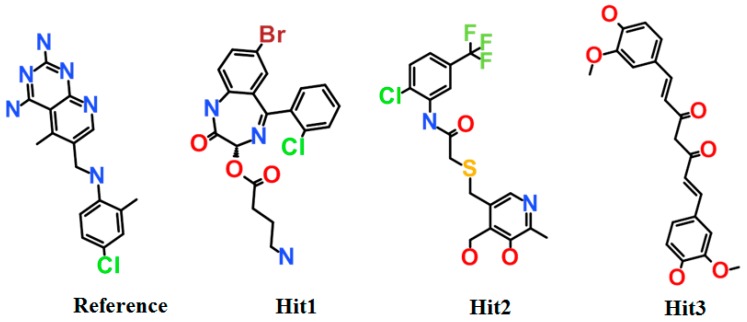
Dihydrofolate reductase (DHFR) is an essential cellular enzyme and thereby catalyzes thereduction of dihydrofolate to tetrahydrofolate (THF). In cancer medication, inhibition of humanDHFR (hDHFR) remains a promising strategy, as it depletes THF and slows DNA synthesis and cellproliferation. In the current study, ligand-based pharmacophore modeling identified and evaluatedthe critical chemical features of hDHFR inhibitors. A pharmacophore model (Hypo1) was generatedfrom known inhibitors of DHFR with a correlation coefficient (0.94), root mean square (RMS)deviation (0.99), and total cost value (125.28). Hypo1 was comprised of four chemical features,including two hydrogen bond donors (HDB), one hydrogen bond acceptor (HBA), and onehydrophobic (HYP). Hypo1 was validated using Fischer's randomization, test set, and decoy setvalidations, employed as a 3D query in a virtual screening at Maybridge, Chembridge, Asinex,National Cancer Institute (NCI), and Zinc databases. Hypo1-retrieved compounds were filtered byan absorption, distribution, metabolism, excretion, and toxicity (ADMET) assessment test andLipinski's rule of five, where the drug-like hit compounds were identified. The hit compounds weredocked in the active site of hDHFR and compounds with Goldfitness score was greater than 44.67(docking score for the reference compound), clustering analysis, and hydrogen bond interactionswere identified. Furthermore, molecular dynamics (MD) simulation identified three compounds asthe best inhibitors of hDHFR with the lowest root mean square deviation (1.2 Å to 1.8 Å), hydrogenbond interactions with hDHFR, and low binding free energy (-127 kJ/mol to -178 kJ/mol). Finally,the toxicity prediction by computer (TOPKAT) affirmed the safety of the novel inhibitors of hDHFRin human body. Overall, we recommend novel hit compounds of hDHFR for cancer and rheumatoidarthritis chemotherapeutics.
二氢叶酸还原酶(DHFR)是一种重要的细胞酶,可催化二氢叶酸还原为四氢叶酸(THF)。在癌症治疗中,抑制人二氢叶酸还原酶(hDHFR)仍然是一种有前景的策略,因为它会耗尽THF并减缓DNA合成和细胞增殖。在当前的研究中,基于配体的药效团建模确定并评估了hDHFR抑制剂的关键化学特征。从已知的DHFR抑制剂生成了一个药效团模型(Hypo1),其相关系数为0.94,均方根(RMS)偏差为0.99,总成本值为125.28。Hypo1由四个化学特征组成,包括两个氢键供体(HDB)、一个氢键受体(HBA)和一个疏水基团(HYP)。通过费舍尔随机化、测试集和诱饵集验证对Hypo1进行了验证,并将其用作在Maybridge、Chembridge、Asinex、美国国立癌症研究所(NCI)和Zinc数据库中进行虚拟筛选的3D查询。通过吸收、分布、代谢、排泄和毒性(ADMET)评估测试以及Lipinski的五规则对Hypo1检索到的化合物进行筛选,从而鉴定出类药物的命中化合物。将命中化合物对接至hDHFR的活性位点,并鉴定出Goldfitness评分大于44.67(参考化合物的对接分数)的化合物、聚类分析和氢键相互作用。此外,分子动力学(MD)模拟确定了三种化合物是hDHFR的最佳抑制剂,其均方根偏差最低(1.2 Å至1.8 Å),与hDHFR有氢键相互作用,且结合自由能较低(-127 kJ/mol至-178 kJ/mol)。最后,计算机毒性预测(TOPKAT)证实了hDHFR新型抑制剂在人体中的安全性。总体而言,我们推荐hDHFR的新型命中化合物用于癌症和类风湿性关节炎的化疗。