Molecular Biology Laboratory, Fundació Puigvert, Instituto de Investigaciones Biomédicas Sant Pau (IIB-Sant Pau), Universitat Autònoma de Barcelona, REDinREN, Instituto de Investigación Carlos III, Cartagena 340-350, 08025, Barcelona, Catalonia, Spain.
Nephrology Department, Fundació Puigvert, Instituto de Investigaciones Biomédicas Sant Pau (IIB-Sant Pau), Universitat Autònoma de Barcelona, REDinREN, Instituto de Investigación Carlos III, Barcelona, Catalonia, Spain.
BMC Nephrol. 2019 Apr 11;20(1):126. doi: 10.1186/s12882-019-1317-y.
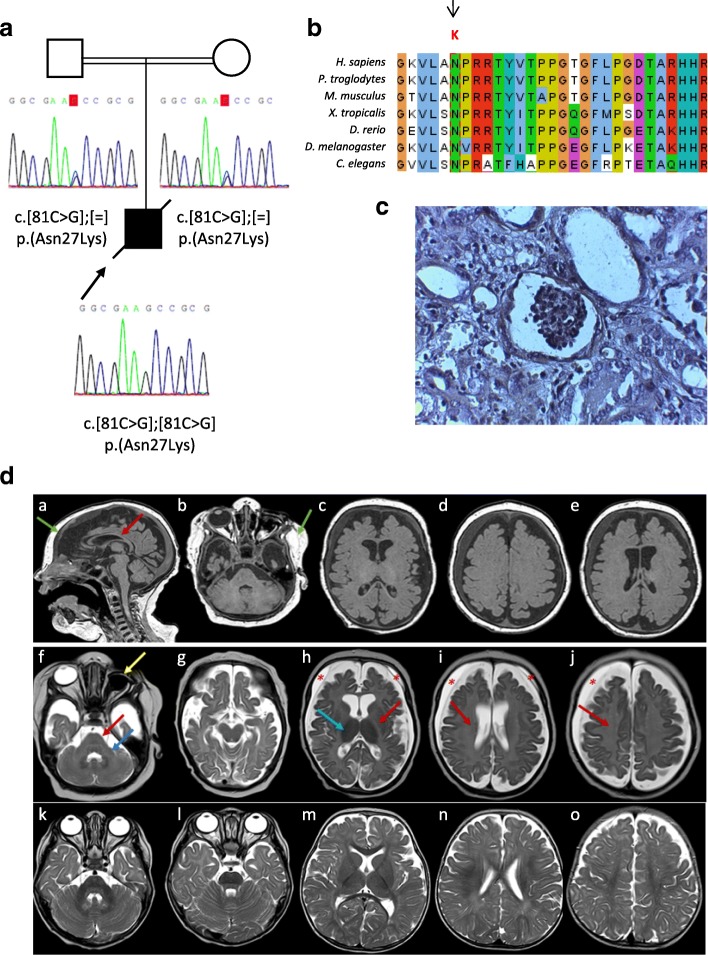
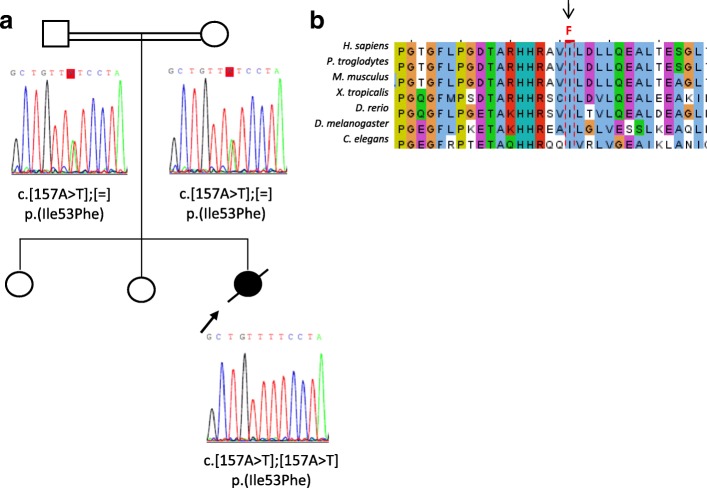
Galloway-Mowat syndrome (GAMOS) is a rare autosomal recessive disorder characterized by early-onset nephrotic syndrome and microcephaly with brain anomalies. WDR73 pathogenic variants were described as the first genetic cause of GAMOS and, very recently, four novel causative genes, OSGEP, LAGE3, TP53RK, and TPRKB, have been identified.
We present the clinical and genetic characteristics of two unrelated infants with clinical suspicion of GAMOS who were born from consanguineous parents. Both patients showed a similar clinical presentation, with early-onset nephrotic syndrome, microcephaly, brain atrophy, developmental delay, axial hypotonia, and early fatality. We identified two novel likely disease-causing variants in the OSGEP gene. These two cases, in conjunction with the findings of a literature review, indicate that OSGEP pathogenic variants are associated with an earlier onset of nephrotic syndrome and shorter life expectancy than WDR73 pathogenic variants.
Our findings expand the spectrum of pathogenic variants in the OSGEP gene and, taken in conjunction with the results of the literature review, suggest that the OSGEP gene should be considered the main known monogenic cause of GAMOS. Early genetic diagnosis of GAMOS is of paramount importance for genetic counseling and family planning.
Galloway-Mowat 综合征(GAMOS)是一种罕见的常染色体隐性遗传病,其特征为早发性肾病综合征和小头畸形伴脑异常。WDR73 致病性变异被描述为 GAMOS 的第一个遗传原因,最近又发现了四个新的致病基因,即 OSGEP、LAGE3、TP53RK 和 TPRKB。
我们介绍了两例来自近亲的临床疑似 GAMOS 的婴儿的临床和遗传特征。两名患者表现出相似的临床表现,即早发性肾病综合征、小头畸形、脑萎缩、发育迟缓、轴向张力减退和早期死亡。我们在 OSGEP 基因中发现了两个新的可能致病变异。这两个病例与文献复习结果一起表明,OSGEP 致病性变异与 WDR73 致病性变异相比,肾病综合征的发病更早,预期寿命更短。
我们的发现扩展了 OSGEP 基因的致病性变异谱,结合文献复习结果,提示 OSGEP 基因应被视为 GAMOS 的主要已知单基因病因。对 GAMOS 进行早期遗传诊断对于遗传咨询和计划生育至关重要。