Harry Perkins Institute of Medical Research, QEII Medical Centre and Centre for Medical Research, The University of Western Australia, Nedlands, Perth, WA 6009, Australia.
Bioinformatics. 2019 Nov 1;35(22):4688-4695. doi: 10.1093/bioinformatics/btz292.
Single-cell RNA sequencing (scRNA-seq) measures gene expression at the resolution of individual cells. Massively multiplexed single-cell profiling has enabled large-scale transcriptional analyses of thousands of cells in complex tissues. In most cases, the true identity of individual cells is unknown and needs to be inferred from the transcriptomic data. Existing methods typically cluster (group) cells based on similarities of their gene expression profiles and assign the same identity to all cells within each cluster using the averaged expression levels. However, scRNA-seq experiments typically produce low-coverage sequencing data for each cell, which hinders the clustering process.
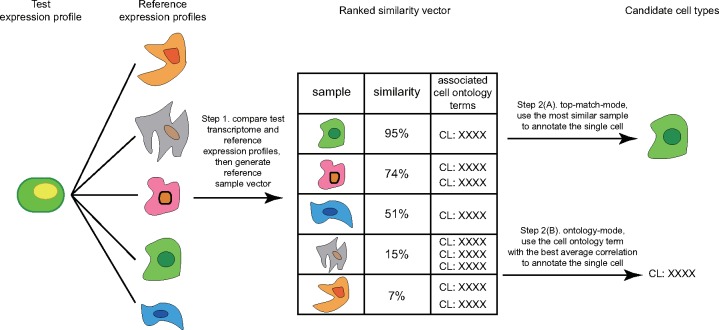
We introduce scMatch, which directly annotates single cells by identifying their closest match in large reference datasets. We used this strategy to annotate various single-cell datasets and evaluated the impacts of sequencing depth, similarity metric and reference datasets. We found that scMatch can rapidly and robustly annotate single cells with comparable accuracy to another recent cell annotation tool (SingleR), but that it is quicker and can handle larger reference datasets. We demonstrate how scMatch can handle large customized reference gene expression profiles that combine data from multiple sources, thus empowering researchers to identify cell populations in any complex tissue with the desired precision.
scMatch (Python code) and the FANTOM5 reference dataset are freely available to the research community here https://github.com/forrest-lab/scMatch.
Supplementary data are available at Bioinformatics online.
单细胞 RNA 测序 (scRNA-seq) 以单个细胞的分辨率测量基因表达。大规模多重单细胞分析使对复杂组织中数千个细胞的大规模转录分析成为可能。在大多数情况下,单个细胞的真实身份未知,需要从转录组数据中推断出来。现有的方法通常根据基因表达谱的相似性对细胞进行聚类(分组),并使用每个聚类中所有细胞的平均表达水平为每个聚类分配相同的身份。然而,scRNA-seq 实验通常为每个细胞产生低覆盖测序数据,这阻碍了聚类过程。
我们引入了 scMatch,它通过在大型参考数据集内识别最接近的匹配来直接注释单细胞。我们使用这种策略注释了各种单细胞数据集,并评估了测序深度、相似性度量和参考数据集的影响。我们发现 scMatch 可以快速而稳健地注释单细胞,其准确性可与另一个最近的细胞注释工具(SingleR)相媲美,但速度更快,并且可以处理更大的参考数据集。我们展示了 scMatch 如何处理大型自定义参考基因表达谱,这些谱结合了来自多个来源的数据,从而使研究人员能够以所需的精度识别任何复杂组织中的细胞群体。
scMatch(Python 代码)和 FANTOM5 参考数据集可在此处免费提供给研究界:https://github.com/forrest-lab/scMatch。
补充数据可在生物信息学在线获得。