Genomics Research Center, AbbVie, North Chicago, IL 60064, USA.
Department of Population Health Sciences, Augusta University, Augusta, GA 30912, USA.
Genes (Basel). 2019 Sep 17;10(9):721. doi: 10.3390/genes10090721.
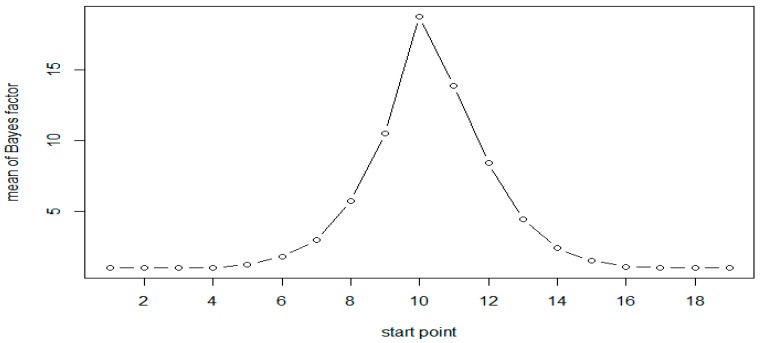
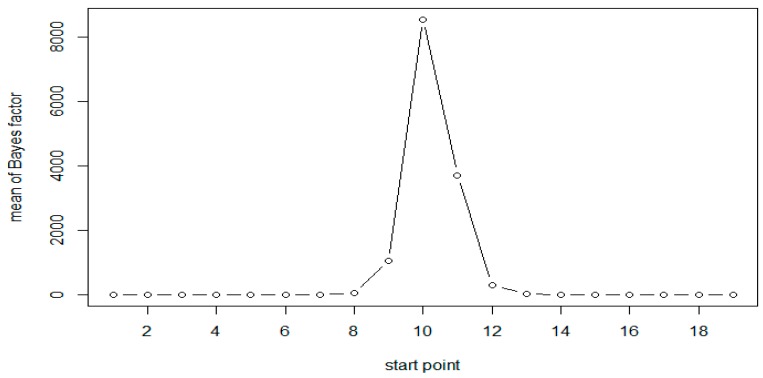
Researchers in genomics are increasingly interested in epigenetic factors such as DNA methylation, because they play an important role in regulating gene expression without changes in the DNA sequence. There have been significant advances in developing statistical methods to detect differentially methylated regions (DMRs) associated with binary disease status. Most of these methods are being developed for detecting differential methylation rates between cases and controls. We consider multiple severity levels of disease, and develop a Bayesian statistical method to detect the region with increasing (or decreasing) methylation rates as the disease severity increases. Patients are classified into more than two groups, based on the disease severity (e.g., stages of cancer), and DMRs are detected by using moving windows along the genome. Within each window, the Bayes factor is calculated to test the hypothesis of monotonic increase in methylation rates corresponding to severity of the disease versus no difference. A mixed-effect model is used to incorporate the correlation of methylation rates of nearby CpG sites in the region. Results from extensive simulation indicate that our proposed method is statistically valid and reasonably powerful. We demonstrate our approach on a bisulfite sequencing dataset from a chronic lymphocytic leukemia (CLL) study.
基因组学研究人员越来越关注表观遗传因素,如 DNA 甲基化,因为它们在调节基因表达方面发挥着重要作用,而不会改变 DNA 序列。在开发用于检测与二元疾病状态相关的差异甲基化区域(DMR)的统计方法方面已经取得了重大进展。这些方法中的大多数都是为检测病例和对照之间的差异甲基化率而开发的。我们考虑疾病的多个严重程度水平,并开发了一种贝叶斯统计方法来检测随着疾病严重程度增加而甲基化率增加(或减少)的区域。根据疾病的严重程度(例如癌症的阶段),患者被分为两个以上的组,并通过在基因组上移动窗口来检测 DMR。在每个窗口中,计算贝叶斯因子以检验与疾病严重程度相比无差异的甲基化率单调增加的假设。使用混合效应模型来合并区域中附近 CpG 位点的甲基化率的相关性。广泛的模拟结果表明,我们提出的方法在统计学上是有效的,并且具有相当的功效。我们在慢性淋巴细胞白血病(CLL)研究的亚硫酸氢盐测序数据集中展示了我们的方法。