Wang Y, Zhao X, Liu-Bryan R
VA San Diego Healthcare System, San Diego, CA, United States; Dept. of Medicine, University of California San Diego, La Jolla, CA, United States.
VA San Diego Healthcare System, San Diego, CA, United States.
Osteoarthritis Cartilage. 2020 May;28(5):669-674. doi: 10.1016/j.joca.2020.01.011. Epub 2020 Jan 30.
Toll-like receptor (TLR)-mediated catabolic responses are implicated to contribute to osteoarthritis (OA). However, deficiency of TLRs has little chondroprotection in mice in vivo. Here, we studied the effect of deficiency of TLR2 and TLR4 in articular chondrocytes on cellular stress responses in vitro.
Chondrocytes isolated from TLR2 and TLR4 double knockout (TLR2/4dKO) and wild type (WT) mice and recombinant HMGB1 (rHMGB1) and LPS were used. Expression of anti-oxidant and DNA repair enzymes including SOD1, SOD2 and OGG1, and phosphorylation of H2AX (a marker for DNA damage) were examined by Western blotting. MitoSOX Red staining was used for assessing mitochondrial superoxide generation. Autophagic activity was monitored by flow cytometry analysis of mean fluorescence intensity (MFI) of GFP and RFP in chondrocytes transfected with a tandem GFP-mRFP-LC3 plasmid, and by Western blot analysis of expression of LC3 and p62, a selective autophagy adaptor.
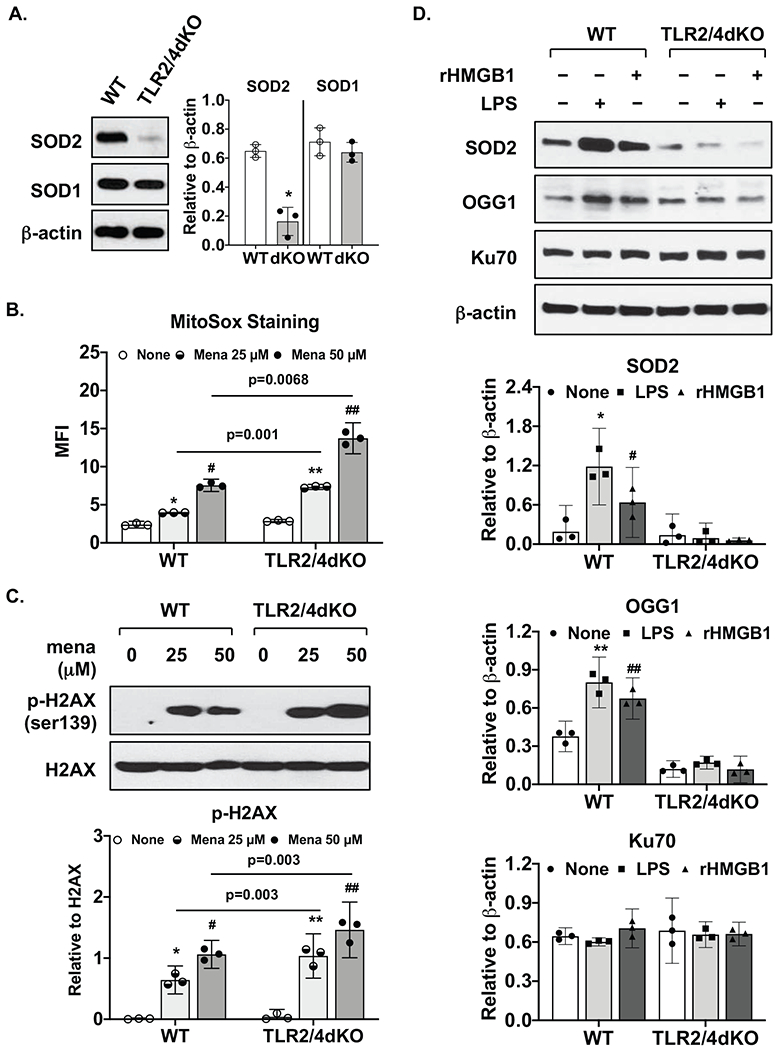
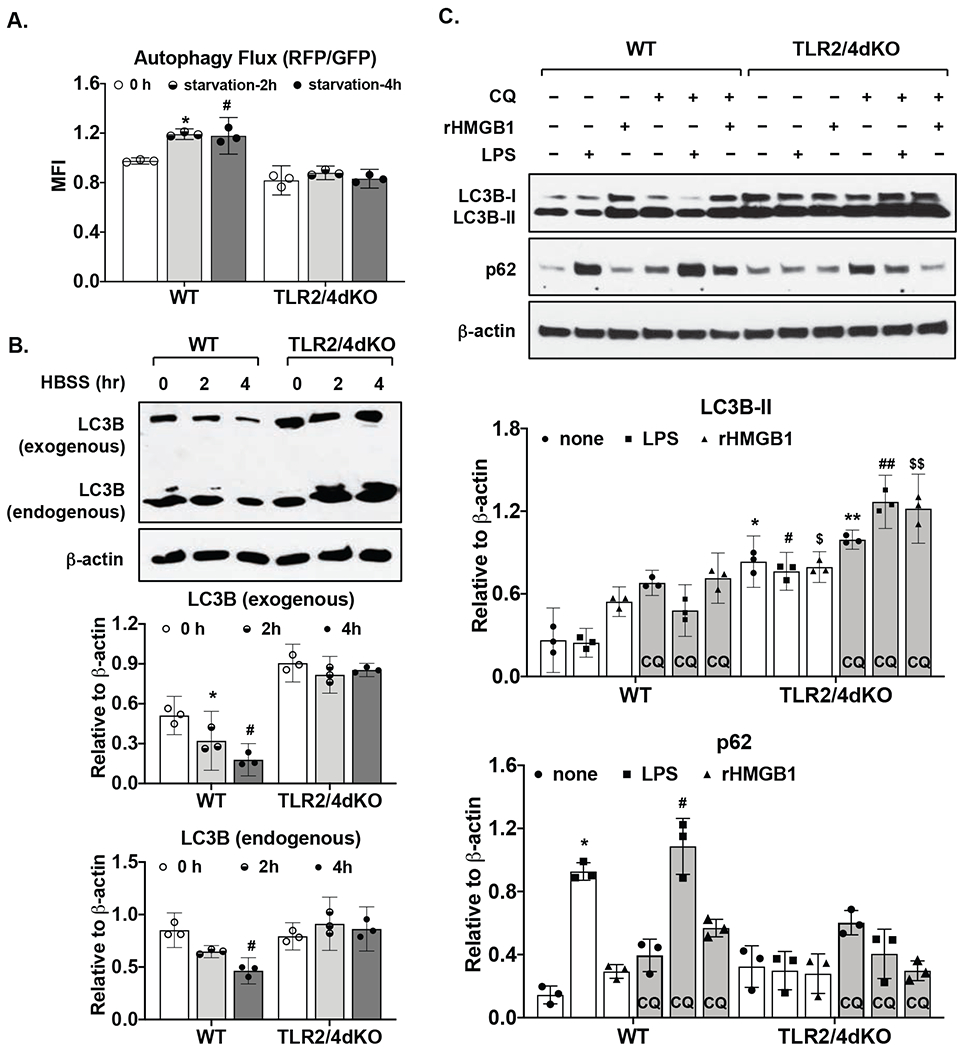
Basal expression of SOD2 but not SOD1 was largely reduced in TLR2/4dKO compared to WT chondrocytes, correlated with significantly enhanced menadione-induced mitochondrial superoxide generation (2.85-3.92 and 3.39 to 8.97 with mean difference 3.39 and 6.18 for 25 and 50μM menadione, respectively) and phosphorylation of H2AX. LPS and rHMGB1 induced expression of SOD2, OGG1 and p62 in WT but not TLR2/4dKO chondrocytes. Autophagy flux was impaired in TLR2/4dKO chondrocytes after acute nutrient stress and by LPS and rHMGB1.
TLR2 and TLR4 deficiency appears to reduce chondrocyte anti-oxidative stress and autophagy flux capacity, which may compromise cartilage homeostasis as a result of chondrocyte dysfunction.
Toll样受体(TLR)介导的分解代谢反应被认为与骨关节炎(OA)的发生有关。然而,在体内小鼠中TLR缺陷对软骨几乎没有保护作用。在此,我们研究了关节软骨细胞中TLR2和TLR4缺陷对体外细胞应激反应的影响。
使用从TLR2和TLR4双敲除(TLR2/4dKO)和野生型(WT)小鼠分离的软骨细胞以及重组高迁移率族蛋白B1(rHMGB1)和脂多糖(LPS)。通过蛋白质免疫印迹法检测抗氧化和DNA修复酶包括超氧化物歧化酶1(SOD1)、超氧化物歧化酶2(SOD2)和8-羟基鸟嘌呤糖苷酶1(OGG1)的表达以及H2AX的磷酸化(DNA损伤的标志物)。使用MitoSOX Red染色评估线粒体超氧化物的产生。通过对转染了串联绿色荧光蛋白-红色荧光蛋白-微管相关蛋白1轻链3(GFP-mRFP-LC3)质粒的软骨细胞中绿色荧光蛋白(GFP)和红色荧光蛋白(RFP)平均荧光强度(MFI)的流式细胞术分析以及通过蛋白质免疫印迹法分析LC3和p62(一种选择性自噬衔接蛋白)的表达来监测自噬活性。
与WT软骨细胞相比,TLR2/4dKO软骨细胞中SOD2的基础表达大幅降低,但SOD1没有,这与甲萘醌诱导的线粒体超氧化物生成显著增强(对于25μM和50μM甲萘醌,分别为2.85 - 3.92和3.39至8.97,平均差异分别为3.39和6.18)以及H2AX的磷酸化相关。LPS和rHMGB1在WT软骨细胞中诱导SOD2、OGG1和p62的表达,但在TLR2/4dKO软骨细胞中未诱导。在急性营养应激后以及LPS和rHMGB1作用下,TLR2/4dKO软骨细胞中的自噬通量受损。
TLR2和TLR4缺陷似乎会降低软骨细胞的抗氧化应激和自噬通量能力,这可能由于软骨细胞功能障碍而损害软骨内环境稳定。