Wang Xi, Han Lin, Wang Xiao-Yan, Wang Jian-Hong, Li Xiao-Meng, Jin Chun-Hua, Wang Lin
Department of Preventive Health Care, Children's Hospital, Capital Institute of Pediatrics, Beijing, China.
Running Gene Inc., Beijing, China.
Front Genet. 2020 Feb 27;11:168. doi: 10.3389/fgene.2020.00168. eCollection 2020.
This study reports a Chinese patient with a Congenital Disorder of Glycosylation (CDG) caused by compound-heterozygous mutations in the Conserved Oligomeric Golgi 5 () gene and thereby offers concrete evidence for early diagnosis.
The clinical manifestations, the results of laboratory examinations and genetic analysis of a 4-year-old Chinese girl with CDG are reported. We also reviewed previous CDG cases that involved mutations by comparing the phenotypes and genotypes in different cases.
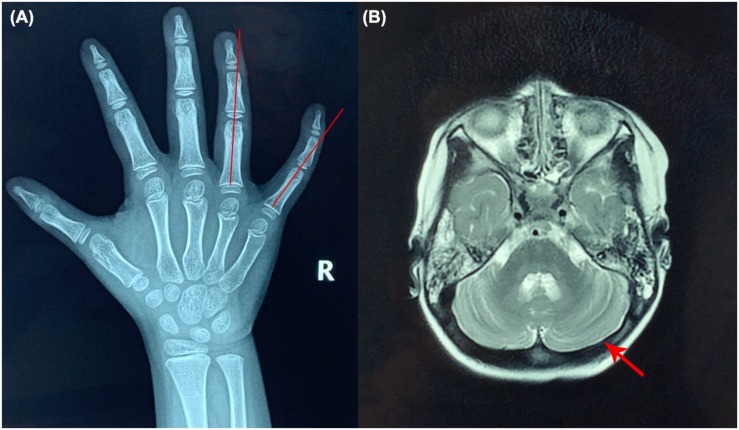
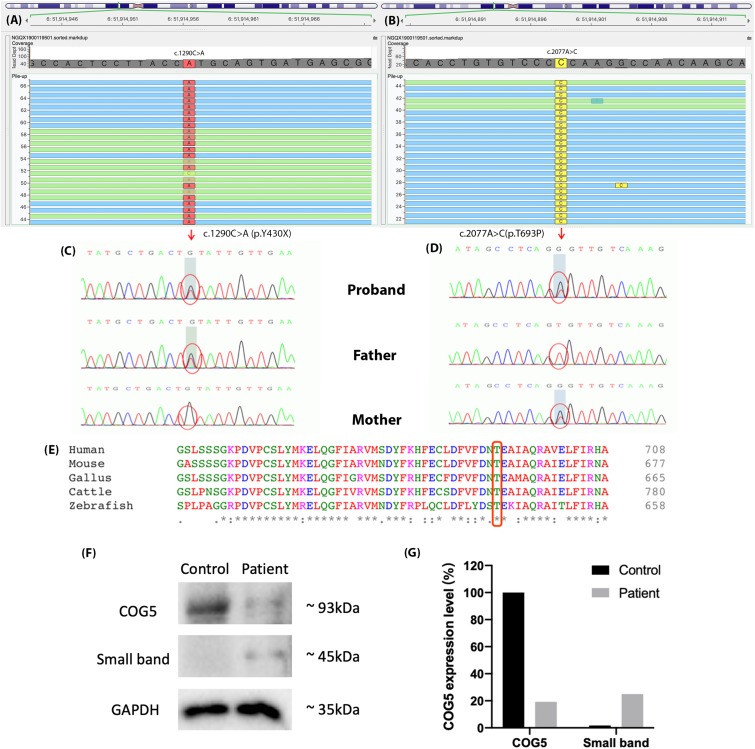
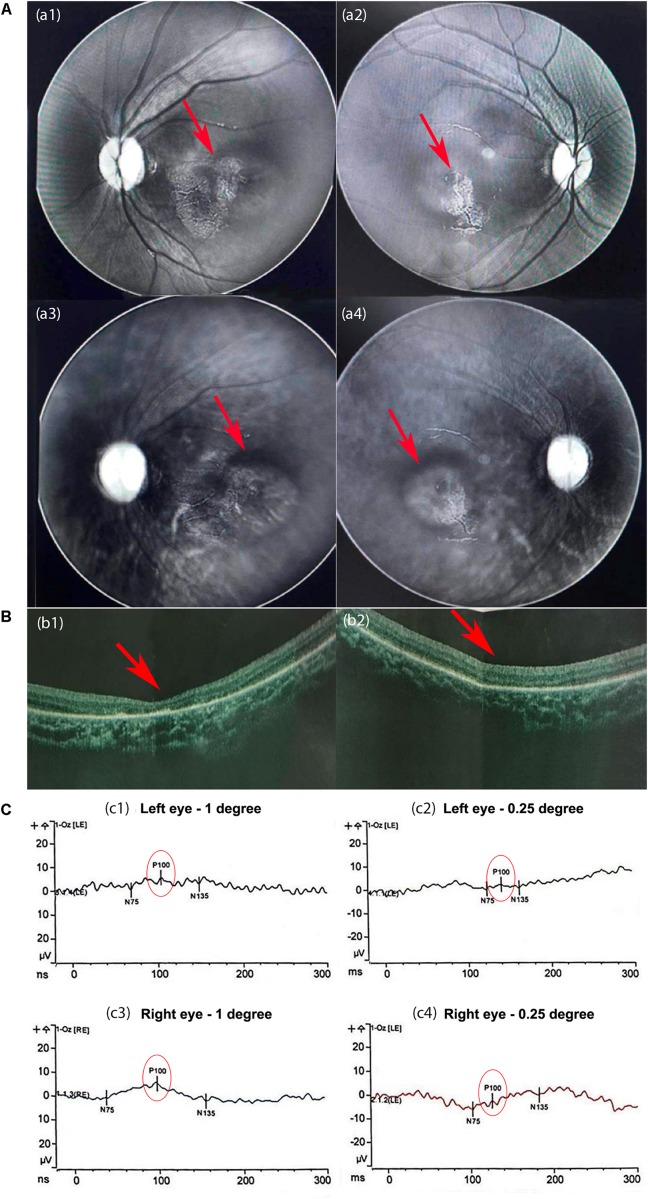
The patient was admitted to our hospital due to ataxia and psychomotor delay. The major clinical manifestations were postural instability, difficulty in walking, psychomotor delay, hypohidrosis, hyperkeratosis of the skin, and ulnar deviation of the right-hand fingers. Biochemical analyses revealed coagulation defect and liver lesions. Vision tests showed choroidopathy and macular hypoplasia. Whole-exome sequencing identified the hitherto unreported compound-heterozygous mutations, c.1290C > A (p.Y430X) and c.2077A > C (p.T693P). Mutation p.Y430X is nonsense, leading to a truncated protein. Mutation p.T693P is located at a highly conserved region, and thus the polar-to-non-polar substitution presumably affects the structure and function of COG5. According to the Human Genome Mutation Database Professional, there have been totally 13 CDG cases caused by 13 mutations. They are mainly characterized by psychomotor delay, hypotonia, ataxia, microcephaly, and hearing and visual abnormalities.
The clinical manifestations of the patient are mild but consistent with the clinical characteristics of the published COG5-CDG cases. The results of this study extend the spectrum of clinical and genetic findings in COG5-CDG.
本研究报告了一名因保守寡聚高尔基体5(COG5)基因复合杂合突变导致先天性糖基化障碍(CDG)的中国患者,从而为早期诊断提供了具体证据。
报告了一名患有CDG的4岁中国女孩的临床表现、实验室检查结果和基因分析。我们还通过比较不同病例的表型和基因型,回顾了先前涉及COG5突变的CDG病例。
该患者因共济失调和精神运动发育迟缓入院。主要临床表现为姿势不稳、行走困难、精神运动发育迟缓、少汗、皮肤角化过度以及右手手指尺侧偏斜。生化分析显示凝血缺陷和肝脏病变。视力测试显示脉络膜病变和黄斑发育不全。全外显子测序确定了迄今未报道的复合杂合COG5突变,即c.1290C>A(p.Y430X)和c.2077A>C(p.T693P)。p.Y430X突变为无义突变,导致蛋白质截短。p.T693P突变位于一个高度保守区域,因此极性到非极性的取代可能影响COG5的结构和功能。根据人类基因组突变数据库专业版,共有13例由13种COG5突变引起的CDG病例。它们的主要特征是精神运动发育迟缓、肌张力减退、共济失调、小头畸形以及听力和视觉异常。
该患者的临床表现较轻,但与已发表的COG5-CDG病例的临床特征一致。本研究结果扩展了COG5-CDG的临床和基因发现谱。