Sahbani Dalila, Strumbo Bice, Tedeschi Silvana, Conte Elena, Camerino Giulia Maria, Benetti Elisa, Montini Giovanni, Aceto Gabriella, Procino Giuseppe, Imbrici Paola, Liantonio Antonella
Department of Pharmacy-Drug Sciences, University of Bari "Aldo Moro", Bari, Italy.
Laboratory of Medical Genetics, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Front Pharmacol. 2020 Mar 17;11:327. doi: 10.3389/fphar.2020.00327. eCollection 2020.
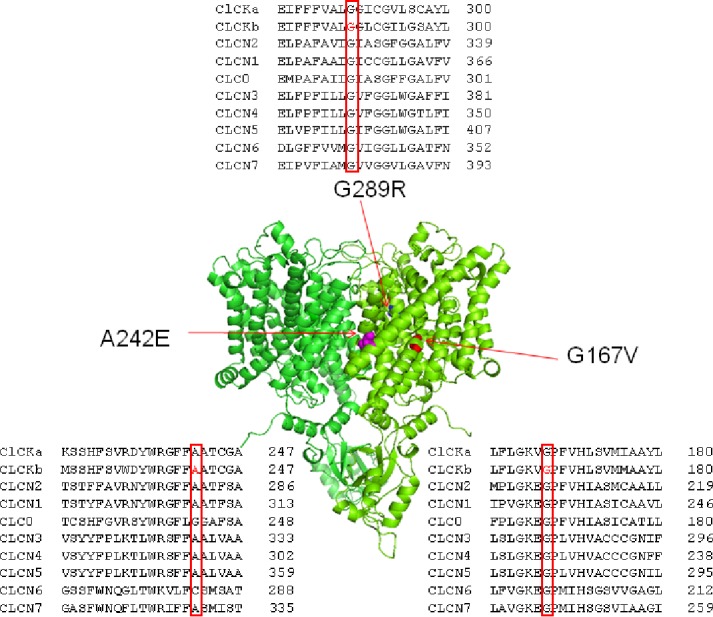
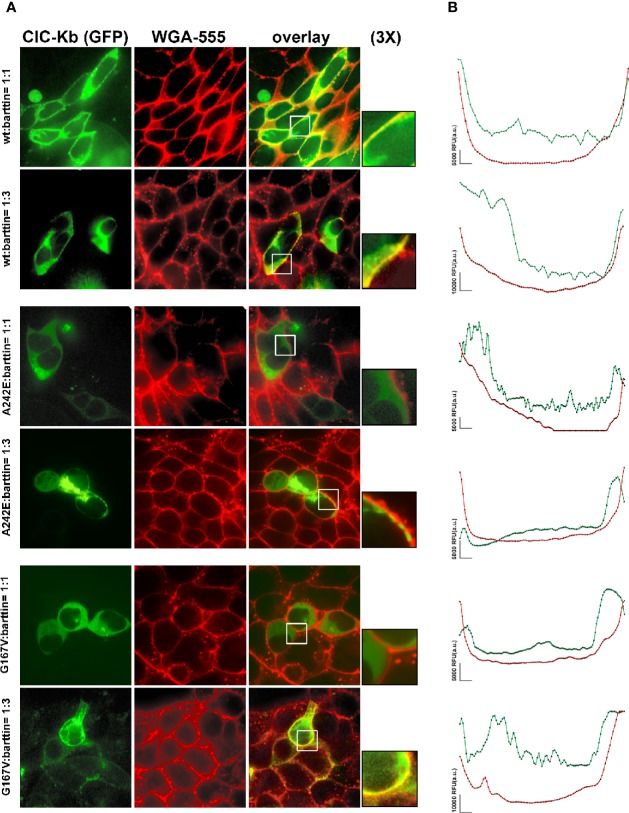
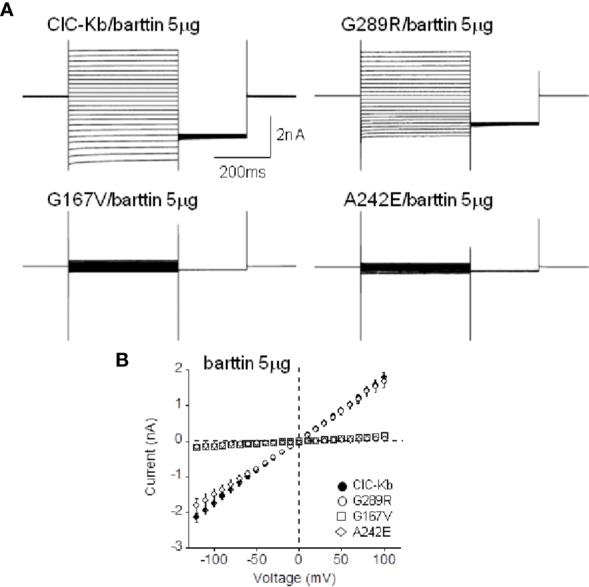
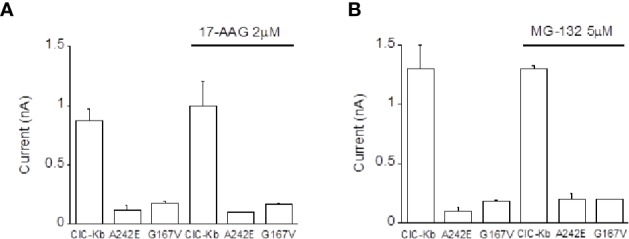
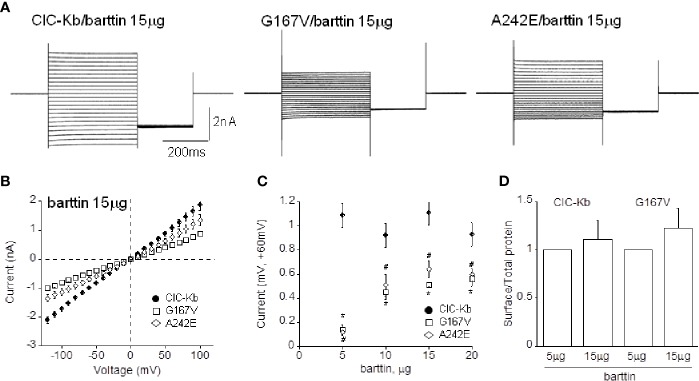
Type III and IV Bartter syndromes (BS) are rare kidney tubulopathies caused by loss-of-function mutations in the and genes coding respectively for the ClC-Kb chloride channels and accessory subunit barttin. ClC-K channels are expressed in the Henle's loop, distal convoluted tubule, and cortical collecting ducts of the kidney and contribute to chloride absorption and urine concentration. In our Italian cohort, we identified two new mutations in , G167V and G289R, in children affected by BS and previously reported genetic variants, A242E, a chimeric gene and the deletion of the whole . All the patients had hypokalemia and metabolic alkalosis, increased serum renin and aldosterone levels and were treated with a symptomatic therapy. In order to define the molecular mechanisms responsible for BS, we co-expressed ClC-Kb wild type and channels with point mutations with barttin in HEK 293 cells and characterized chloride currents through the patch-clamp technique. In addition, we attempted to revert the functional defect caused by BS mutations through barttin overexpression. G167V and A242E channels showed a drastic current reduction compared to wild type, likely suggesting compromised expression of mutant channels at the plasma membrane. Conversely, G289R channel was similar to wild type raising the doubt that an additional mutation in another gene or other mechanisms could account for the clinical phenotype. Interestingly, increasing ClC-K/barttin ratio augmented G167V and A242E mutants' chloride current amplitudes towards wild type levels. These results confirm a genotype-phenotype correlation in BS and represent a preliminary proof of concept that molecules functioning as molecular chaperones can restore channel function in expression-defective ClC-Kb mutants.
III型和IV型巴特综合征(BS)是罕见的肾小管病,由分别编码ClC-Kb氯通道和辅助亚基barttin的基因功能丧失突变引起。ClC-K通道在肾脏的髓袢、远曲小管和皮质集合管中表达,有助于氯的吸收和尿液浓缩。在我们的意大利队列中,我们在受BS影响的儿童中发现了两个新的 突变,即G167V和G289R,以及先前报道的基因变体A242E、一个嵌合基因和整个 的缺失。所有患者均有低钾血症和代谢性碱中毒,血清肾素和醛固酮水平升高,并接受了对症治疗。为了确定导致BS的分子机制,我们在HEK 293细胞中共同表达了ClC-Kb野生型和带有点突变的通道与barttin,并通过膜片钳技术对氯电流进行了表征。此外,我们试图通过barttin过表达来恢复由BS突变引起的功能缺陷。与野生型相比,G167V和A242E通道的电流大幅降低,这可能表明突变通道在质膜上的表达受损。相反,G289R通道与野生型相似,这让人怀疑另一个基因中的额外突变或其他机制可能是临床表型的原因。有趣的是,增加ClC-K/barttin的比例可使G167V和A242E突变体的氯电流幅度向野生型水平增加。这些结果证实了BS中的基因型-表型相关性,并代表了一个初步的概念验证,即作为分子伴侣发挥作用的分子可以恢复表达缺陷的ClC-Kb突变体中的通道功能。