Suzuki Shinya, Yamada Takuji
School of Life Science and Technology, Tokyo Institute of Technology, Meguro, Tokyo, Japan.
PeerJ. 2020 Mar 27;8:e8722. doi: 10.7717/peerj.8722. eCollection 2020.
With the development of DNA sequencing technology, static omics profiling in microbial communities, such as taxonomic and functional gene composition determination, has become possible. Additionally, the recently proposed in situ growth rate estimation method allows the applicable range of current comparative metagenomics to be extended to dynamic profiling. However, with this method, the applicable target range is presently limited. Furthermore, the characteristics of coverage depth during replication have not been sufficiently investigated.
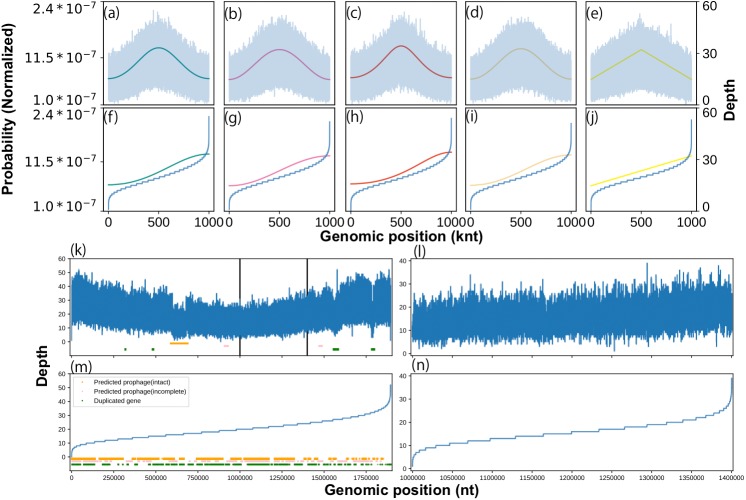
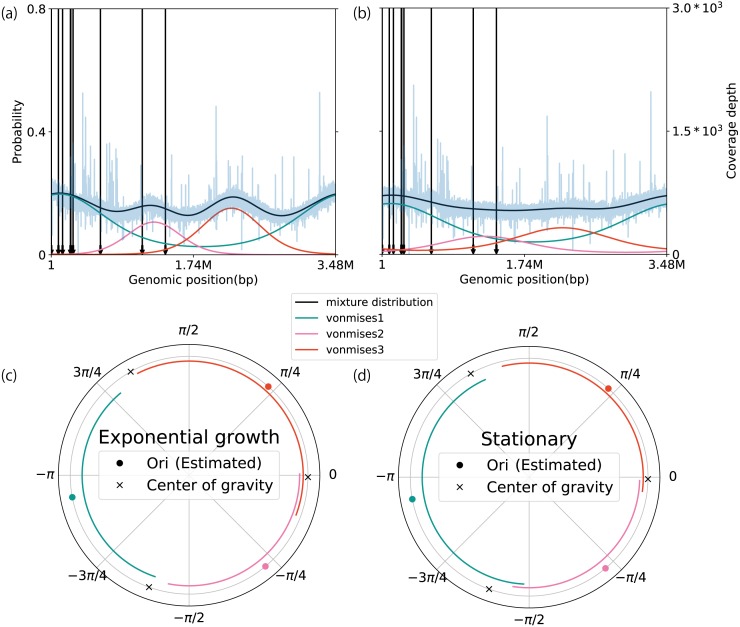
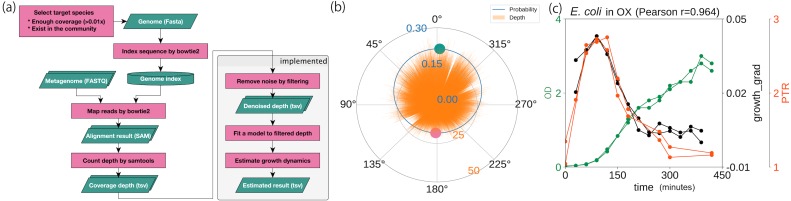
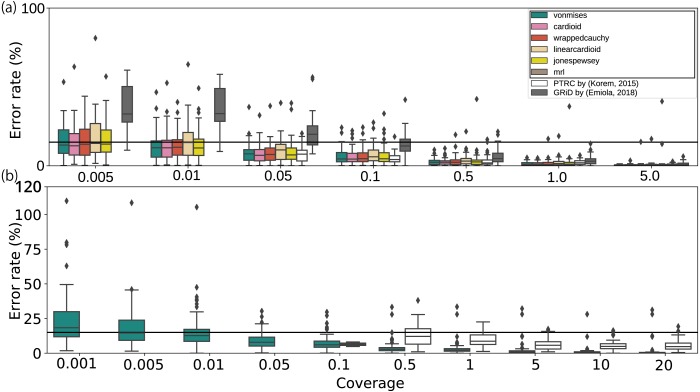
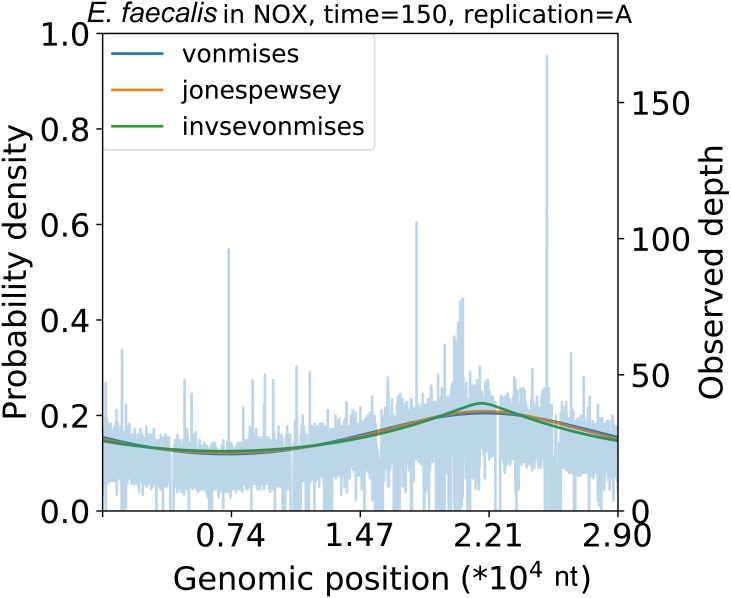
We developed a probabilistic model that mimics coverage depth dynamics. This statistical model explains the bias that occurs in the coverage depth due to DNA replication and errors that arise from coverage depth observation. Although our method requires a complete genome sequence, it involves a stable to low coverage depth (>0.01×). We also evaluated the estimation using real whole-genome sequence datasets and reproduced the growth dynamics observed in previous studies. By utilizing a circular distribution in the model, our method facilitates the quantification of unmeasured coverage depth features, including peakedness, skewness, and degree of density, around the replication origin. When we applied the model to time-series culture samples, the skewness parameter, which indicates the asymmetry, was stable over time; however, the peakedness and degree of density parameters, which indicate the concentration level at the replication origin, changed dynamically. Furthermore, we demonstrated the activity measurement of multiple replication origins in a single chromosome.
We devised a novel framework for quantifying coverage depth dynamics. Our study is expected to serve as a basis for replication activity estimation from a broader perspective using the statistical model.
随着DNA测序技术的发展,对微生物群落进行静态组学分析,如确定分类学和功能基因组成,已成为可能。此外,最近提出的原位生长速率估计方法使当前比较宏基因组学的适用范围扩展到动态分析。然而,使用这种方法,目前适用的目标范围有限。此外,复制过程中覆盖深度的特征尚未得到充分研究。
我们开发了一个模拟覆盖深度动态的概率模型。这个统计模型解释了由于DNA复制导致的覆盖深度偏差以及覆盖深度观察中出现的误差。虽然我们的方法需要完整的基因组序列,但它适用于稳定到低覆盖深度(>0.01×)。我们还使用真实的全基因组序列数据集评估了估计结果,并重现了先前研究中观察到的生长动态。通过在模型中利用圆形分布,我们的方法有助于量化复制起点周围未测量的覆盖深度特征,包括峰度、偏度和密度程度。当我们将该模型应用于时间序列培养样本时,指示不对称性的偏度参数随时间稳定;然而,指示复制起点处浓度水平的峰度和密度程度参数则动态变化。此外,我们展示了单条染色体上多个复制起点的活性测量。
我们设计了一个用于量化覆盖深度动态的新框架。我们的研究有望为使用统计模型从更广泛的角度估计复制活性提供基础。