Department of Surgery, University of South Florida Morsani College of Medicine, Tampa, FL, United States of America.
Department of Biomolecular Research, Boise State University, Boise, ID, United States of America.
PLoS One. 2020 Apr 30;15(4):e0231739. doi: 10.1371/journal.pone.0231739. eCollection 2020.
We previously reported microvascular leakage resulting from fibrinogen-γ chain C-terminal products (γC) occurred via a RhoA-dependent mechanism. The objective of this study was to further elucidate the signaling mechanism by which γC induces endothelial hyperpermeability. Since it is known that γC binds and activates endothelial αvβ3, a transmembrane integrin receptor involved in intracellular signaling mediated by the tyrosine kinases FAK and Src, we hypothesized that γC alters endothelial barrier function by activating the FAK-Src pathway leading to junction dissociation and RhoA driven cytoskeletal stress-fiber formation.
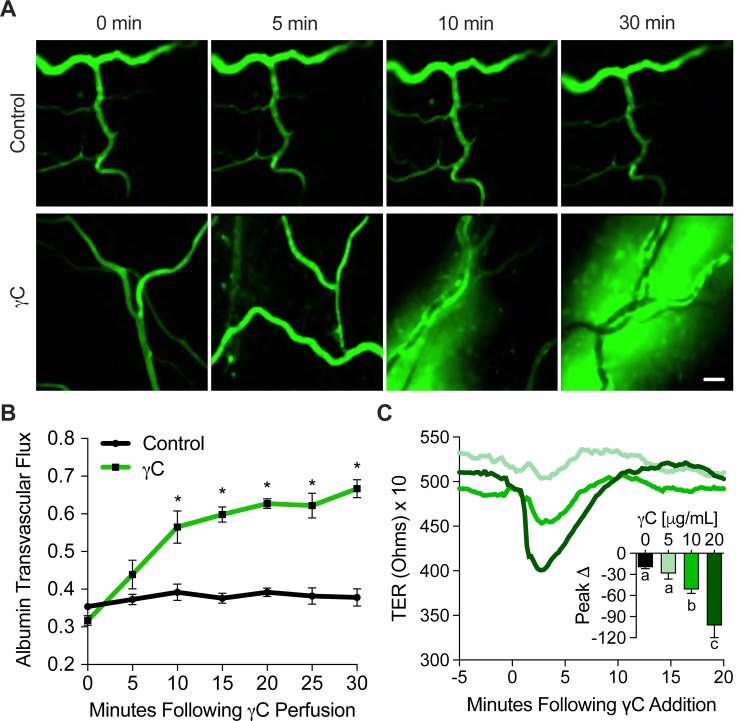
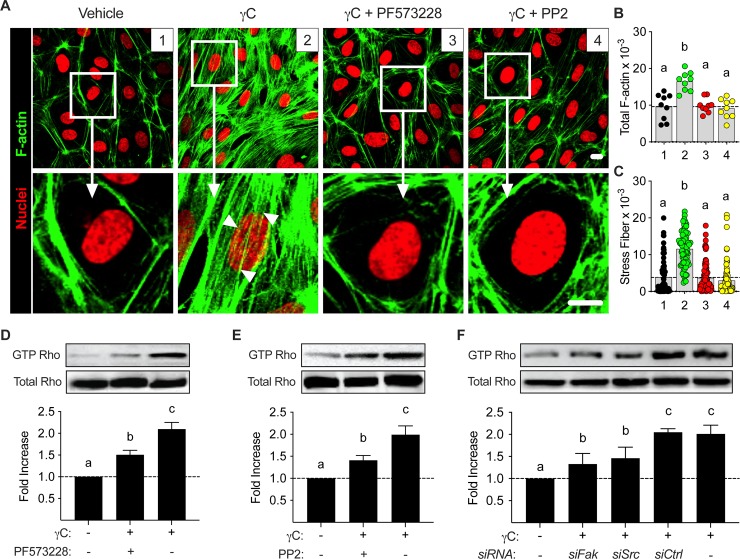
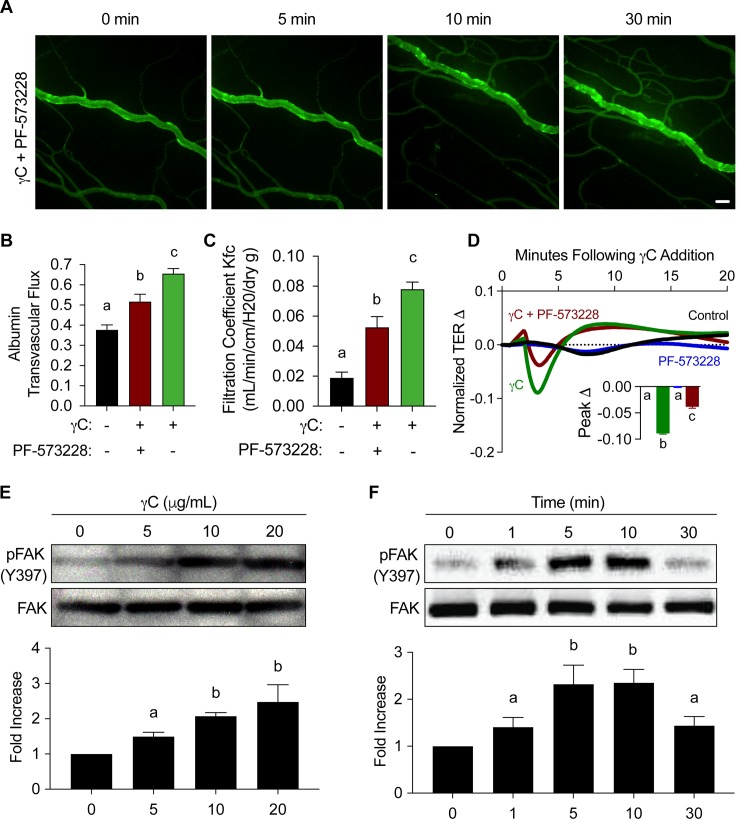
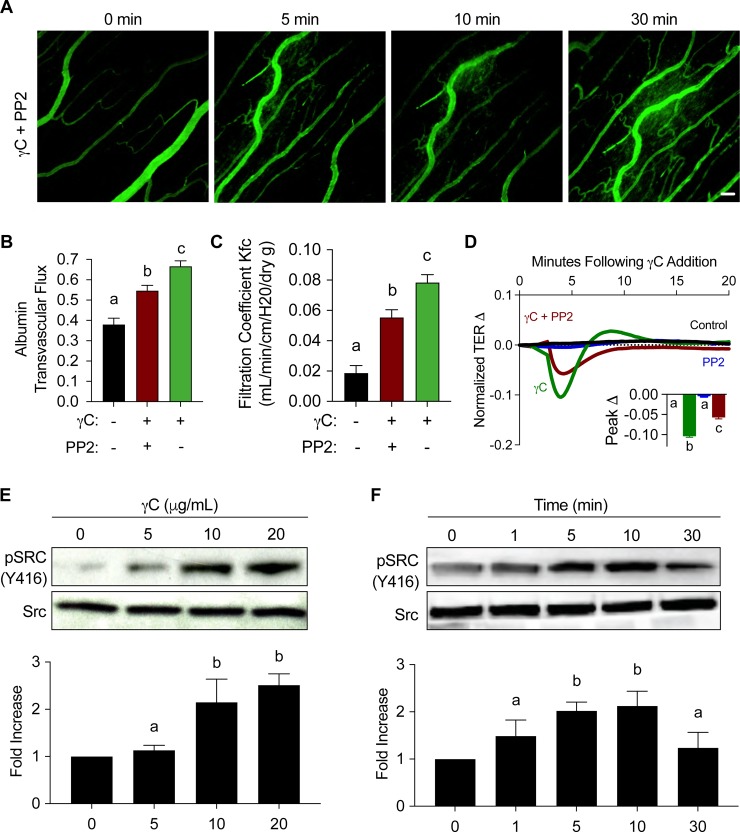
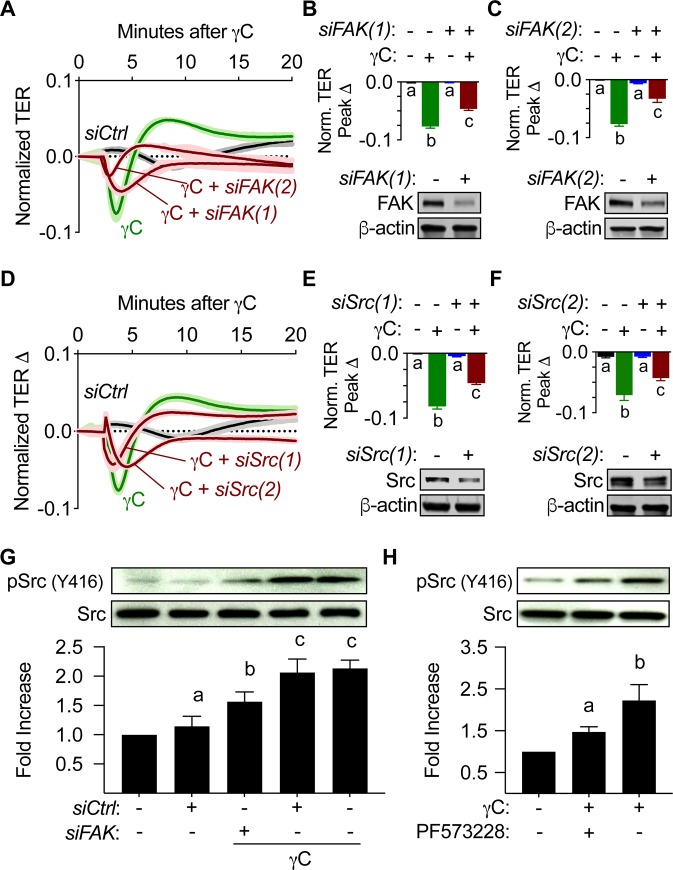
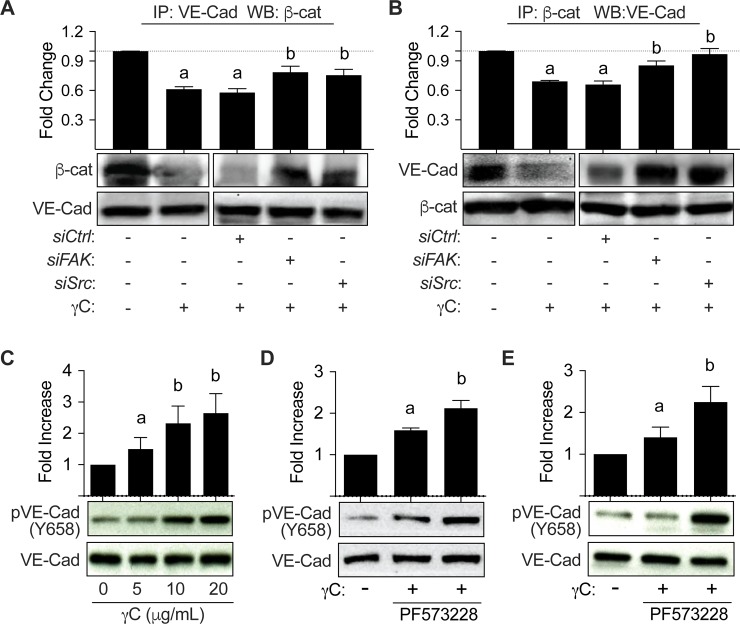
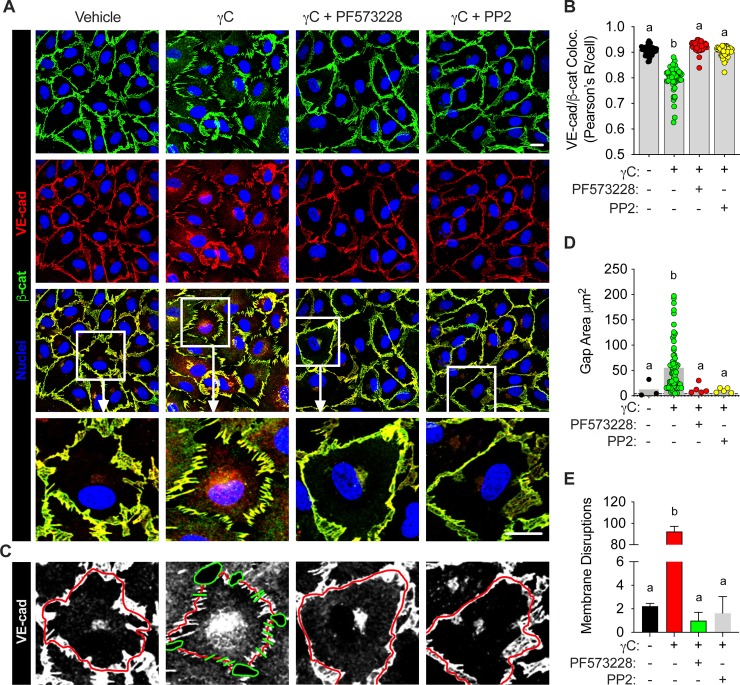
Using intravital microscopy of rat mesenteric microvessels, we show increased extravasation of plasma protein (albumin) resulting from γC administration. In addition, capillary fluid filtration coefficient (Kfc) indicated γC-induced elevated lung vascular permeability. Furthermore, γC decreased transendothelial barrier resistance in a time-dependent and dose-related fashion in cultured rat lung microvascular endothelial cells (RLMVECs), accompanied by increased FAK/Src phosphorylation detection by western blot. Experiments with pharmacological inhibition or gene silencing of FAK showed significantly reduced γC-induced albumin and fluid leakage across microvessels, stress-fiber formation, VE-cadherin tyrosine phosphorylation, and improved γC-induced endothelial barrier dysfunction, indicating the involvement of FAK in γC mediated hyperpermeability. Comparable results were found when Src was targeted in a similar manner, however inhibition of FAK prevented Src activation, suggesting that FAK is upstream of Src in γC-mediated hyperpermeability. In addition, γC-induced cytoskeletal stress-fiber formation was attenuated during inhibition or silencing of these tyrosine kinases, concomitantly with RhoA inhibition.
The FAK-Src pathway contributes to γC-induced microvascular barrier dysfunction, junction protein phosphorylation and disorganization in a manner that involves RhoA and stress-fiber formation.
我们之前报道过纤维蛋白原-γ 链 C 末端产物(γC)导致的微血管渗漏是通过 RhoA 依赖的机制发生的。本研究的目的是进一步阐明 γC 诱导内皮细胞通透性增加的信号转导机制。由于已知 γC 结合并激活参与由酪氨酸激酶 FAK 和 Src 介导的细胞内信号转导的跨膜整合素受体αvβ3,我们假设 γC 通过激活 FAK-Src 途径改变内皮屏障功能,导致连接解离和 RhoA 驱动的细胞骨架应力纤维形成。
使用大鼠肠系膜微血管的活体显微镜检查,我们显示出 γC 给药导致血浆蛋白(白蛋白)的渗出增加。此外,毛细血管流体滤过系数(Kfc)表明 γC 诱导的肺血管通透性增加。此外,γC 以时间依赖性和剂量依赖性方式降低培养的大鼠肺微血管内皮细胞(RLMVEC)中的跨内皮屏障阻力,Western blot 检测到 FAK/Src 磷酸化增加。用药理学抑制或 FAK 基因沉默的实验表明,γC 诱导的白蛋白和流体渗漏、应力纤维形成、VE-钙粘蛋白酪氨酸磷酸化以及改善的 γC 诱导的内皮屏障功能障碍显著减少,表明 FAK 参与了 γC 介导的通透性增加。以类似的方式靶向Src 时也得到了类似的结果,但是抑制 FAK 阻止了 Src 的激活,这表明在 γC 介导的通透性增加中 FAK 位于 Src 的上游。此外,在抑制或沉默这些酪氨酸激酶的同时,γC 诱导的细胞骨架应力纤维形成减弱,同时 RhoA 也被抑制。
FAK-Src 途径参与了 γC 诱导的微血管屏障功能障碍、连接蛋白磷酸化和失稳,涉及 RhoA 和应力纤维形成。