Van Andel Research Institute, Grand Rapids, MI, USA.
Department of Cellular and Molecular Medicine and Prostate Cancer Research Program at University of Arizona Cancer Center, Tucson, AZ, USA.
Oncogene. 2020 Jul;39(31):5390-5404. doi: 10.1038/s41388-020-1370-9. Epub 2020 Jun 21.
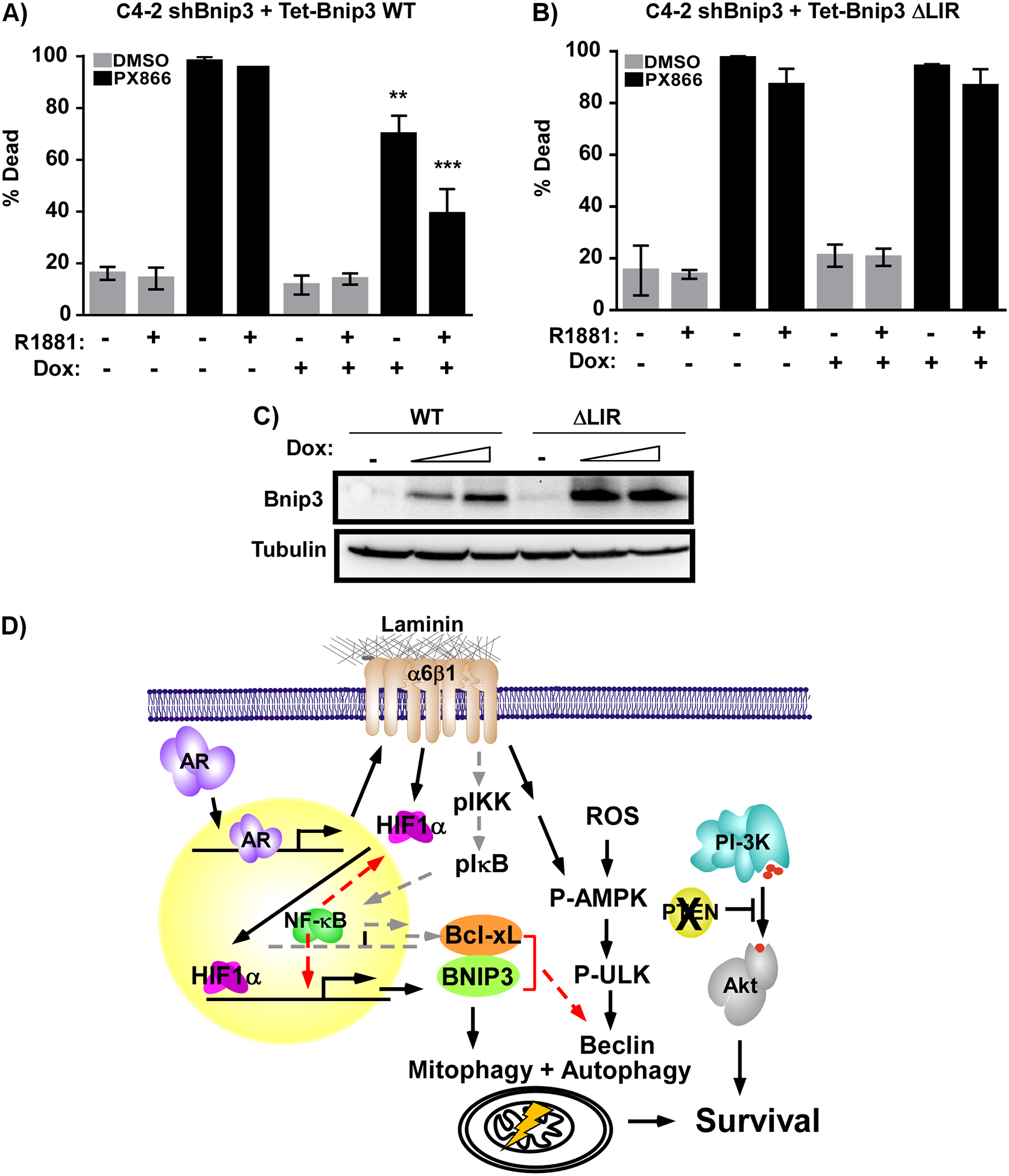
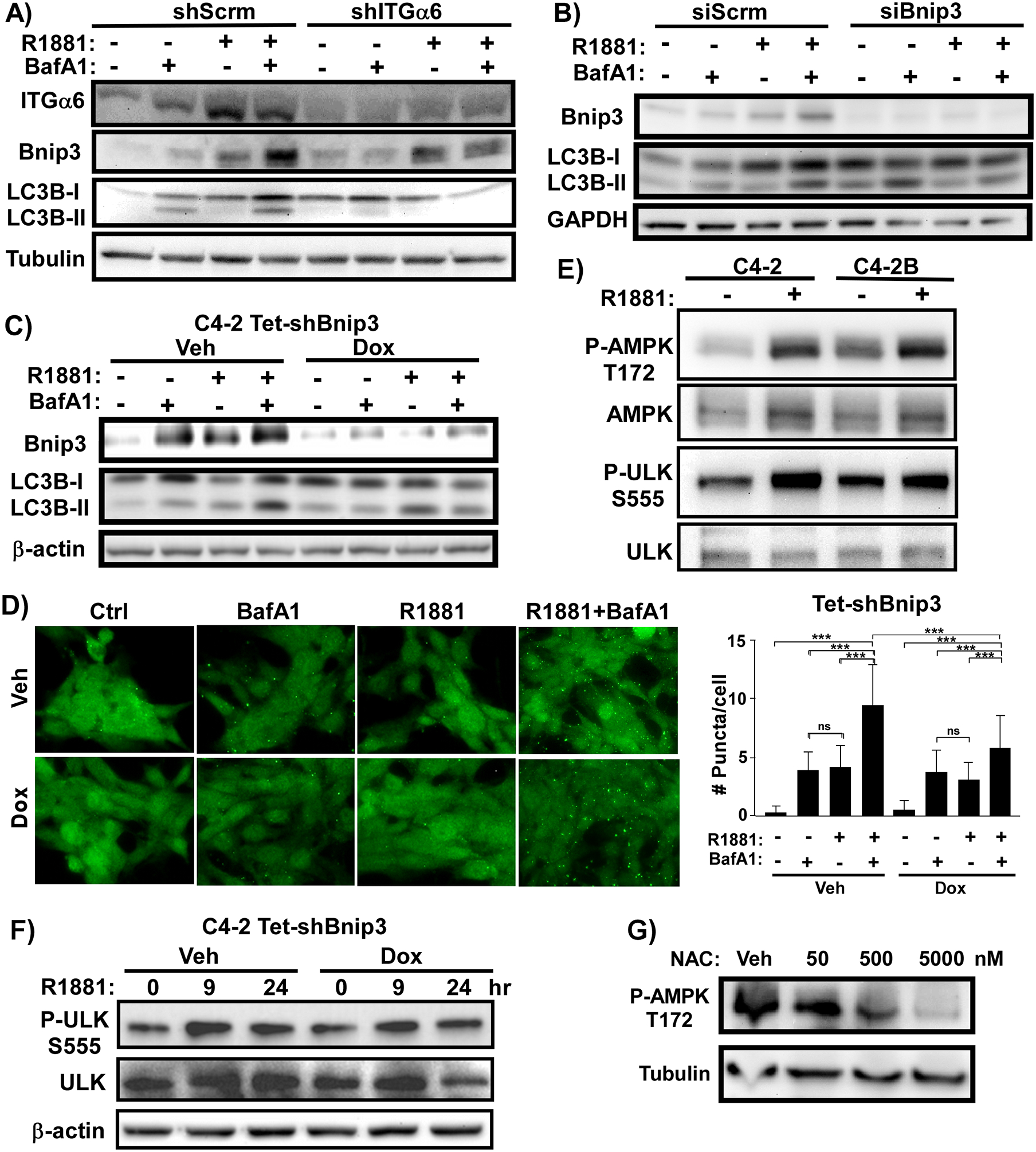
The androgen receptor (AR) is the major driver of prostate cancer growth and survival. However, almost all patients relapse with castration-resistant disease (CRPC) when treated with anti-androgen therapy. In CRPC, AR is often aberrantly activated independent of androgen. Targeting survival pathways downstream of AR could be a viable strategy to overcome CRPC. Surprisingly, little is known about how AR drives prostate cancer survival. Furthermore, CRPC tumors in which Pten is lost are also resistant to eradication by PI3K inhibitors. We sought to identify the mechanism by which AR drives tumor survival in CRPC to identify ways to overcome resistance to PI3K inhibition. We found that integrins α6β1 and Bnip3 are selectively elevated in CRPC downstream of AR. While integrin α6 promotes survival and is a direct transcriptional target of AR, the ability of AR to induce Bnip3 is dependent on adhesion to laminin and integrin α6β1-dependent nuclear translocation of HIF1α. Integrins α6β1 and Bnip3 were found to promote survival of CRPC cells selectively on laminin through the induction of autophagy and mitophagy. Furthermore, blocking Bnip3 or integrin α6β1 restored sensitivity to PI3K inhibitors in Pten-negative CRPC. We identified an AR driven pathway that cooperates with laminin and hypoxia to drive resistance to PI3K inhibitors. These findings can help explain in part why PI3K inhibitors have failed in clinical trials to overcome AR-dependent CRPC.
雄激素受体 (AR) 是前列腺癌生长和存活的主要驱动因素。然而,几乎所有接受抗雄激素治疗的患者在治疗后都会复发去势抵抗性疾病 (CRPC)。在 CRPC 中,AR 通常在没有雄激素的情况下异常激活。靶向 AR 下游的存活途径可能是克服 CRPC 的可行策略。令人惊讶的是,人们对 AR 如何驱动前列腺癌存活知之甚少。此外,丢失 Pten 的 CRPC 肿瘤也对 PI3K 抑制剂的消除具有抗性。我们试图确定 AR 在 CRPC 中驱动肿瘤存活的机制,以确定克服对 PI3K 抑制的耐药性的方法。我们发现整合素 α6β1 和 Bnip3 在 AR 下游的 CRPC 中选择性升高。虽然整合素 α6 促进存活,并且是 AR 的直接转录靶标,但 AR 诱导 Bnip3 的能力依赖于与层粘连蛋白的粘附以及整合素 α6β1 依赖性 HIF1α 的核易位。发现整合素 α6β1 和 Bnip3 通过诱导自噬和线粒体自噬选择性地在层粘连蛋白上促进 CRPC 细胞的存活。此外,阻断 Bnip3 或整合素 α6β1 可恢复对 Pten 阴性 CRPC 中 PI3K 抑制剂的敏感性。我们确定了一种 AR 驱动的途径,该途径与层粘连蛋白和缺氧合作,导致对 PI3K 抑制剂的耐药性。这些发现部分解释了为什么 PI3K 抑制剂在临床试验中未能克服 AR 依赖性 CRPC。