Zhang Hongxi, Li Douyue, Zhao Xiangyan, Pan Saichao, Wu Xiaolong, Peng Shan, Huang Hanrou, Shi Ruixue, Tan Zhongyang
Bioinformatics Center, College of Biology, Hunan University, Changsha, 410082, China.
BMC Genomics. 2020 Aug 17;21(1):563. doi: 10.1186/s12864-020-06949-5.
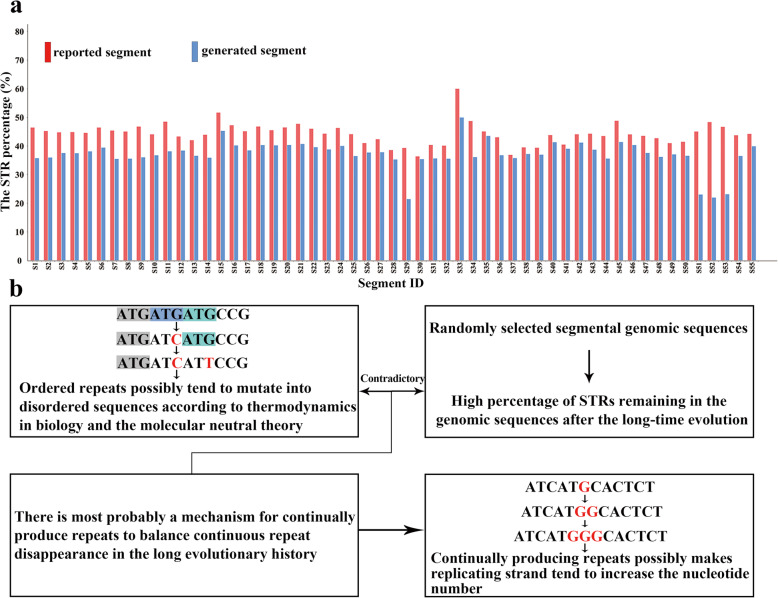
The ubiquitous presence of short tandem repeats (STRs) in virtually all genomes implicates their functional relevance, while a widely-accepted definition of STR is yet to be established. Previous studies majorly focus on relatively longer STRs, while shorter repeats were generally excluded. Herein, we have adopted a more generous criteria to define shorter repeats, which has led to the definition of a much larger number of STRs that lack prior analysis. Using this definition, we analyzed the short repeats in 55 randomly selected segments in 55 randomly selected genomic sequences from a fairly wide range of species covering animals, plants, fungi, protozoa, bacteria, archaea and viruses.
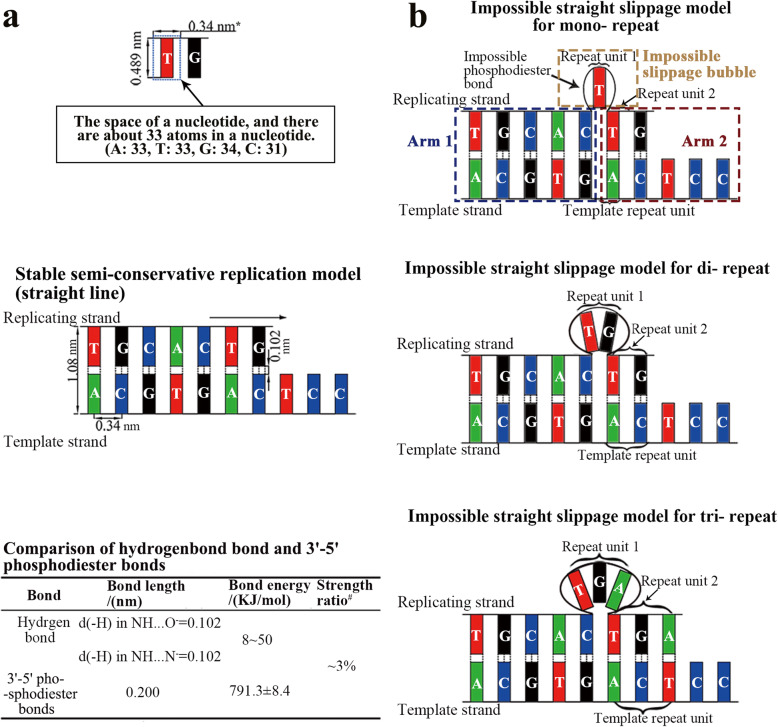
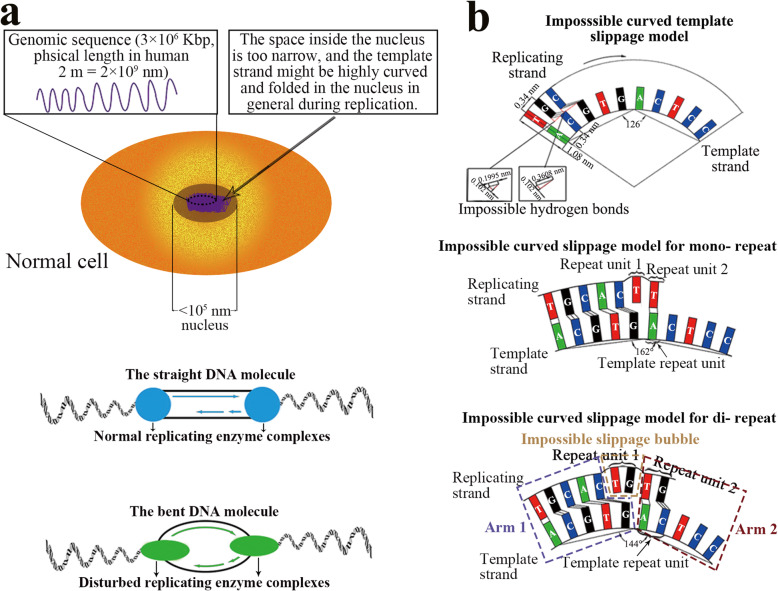
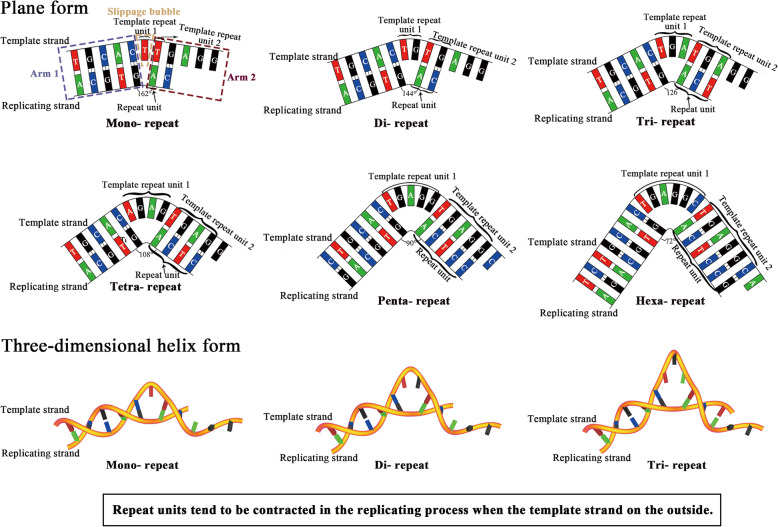
Our analysis reveals a high percentage of short repeats in all 55 randomly selected segments, indicating that the universal presence of high-content short repeats could be a common characteristic of genomes across all biological kingdoms. Therefore, it is reasonable to assume a mechanism for continuous production of repeats that can make the replicating process relatively semi-conservative. We have proposed a folded replication slippage model that considers the geometric space of nucleotides and hydrogen bond stability to explain the mechanism more explicitly, with improving the existing straight-line slippage model. The folded slippage model can explain the expansion and contraction of mono- to hexa- nucleotide repeats with proper folding angles. Analysis of external forces in the folding template strands also suggests that expansion exists more commonly than contraction in the short tandem repeats.
The folded replication slippage model provides a reasonable explanation for the continuous occurrences of simple sequence repeats in genomes. This model also contributes to the explanation of STR-to-genome evolution and is an alternative model that complements semi-conservative replication.
短串联重复序列(STR)几乎普遍存在于所有基因组中,这表明它们具有功能相关性,但尚未建立一个被广泛接受的STR定义。以往的研究主要集中在相对较长的STR上,而较短的重复序列通常被排除在外。在此,我们采用了更宽松的标准来定义较短的重复序列,这导致定义了大量此前未分析过的STR。使用这个定义,我们分析了从动物、植物、真菌、原生动物、细菌、古菌和病毒等相当广泛的物种中随机选择的55个基因组序列中55个随机选择片段中的短重复序列。
我们的分析显示,在所有55个随机选择的片段中,短重复序列的比例很高,这表明高含量短重复序列的普遍存在可能是所有生物界基因组的共同特征。因此,合理假设存在一种连续产生重复序列的机制,使复制过程相对半保守。我们提出了一种折叠复制滑动模型,该模型考虑了核苷酸的几何空间和氢键稳定性,以更明确地解释该机制,改进了现有的直线滑动模型。折叠滑动模型可以用适当的折叠角度解释单核苷酸到六核苷酸重复序列的扩增和收缩。对折叠模板链中外力的分析还表明,在短串联重复序列中,扩增比收缩更常见。
折叠复制滑动模型为基因组中简单序列重复的持续发生提供了合理的解释。该模型也有助于解释STR到基因组的进化,是对半保守复制进行补充的一种替代模型。