Dong Han-Yu, Feng Jun-Yan, Yue Xiao-Jing, Shan Ling, Jia Fei-Yong
Department of Developmental and Behavioral Pediatrics, The First Hospital of Jilin University, Changchun, China.
Medicine (Baltimore). 2020 Aug 14;99(33):e21753. doi: 10.1097/MD.0000000000021753.
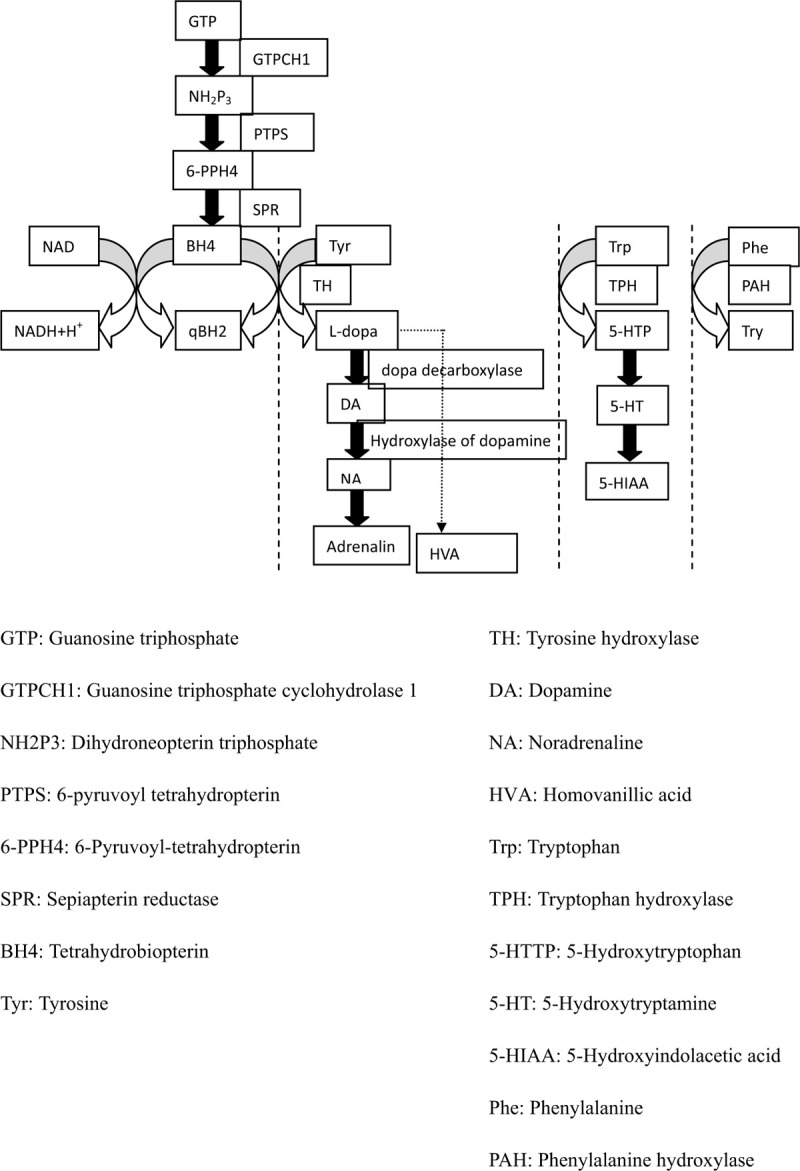
Tyrosine hydroxylase deficiency (THD) is a rare cause of dopa-responsive dystonia (DRD). Although the symptoms of DRD may be improved by treatment with L-dopa, the low morbidity of THD can lead to its misdiagnosis. Thus, it is important for physicians to be aware of THD as a cause of DRD.
We report 3 cases of THD. A 5-year-old boy with DRD was diagnosed with THD and found to have compound heterozygous mutations of the TH gene, including TH:c.647G>C from his mother and TH:c.646G>A from his father. Two female siblings also were found to have TH:c.698G>A from their mother and TH:c.710T>C from their father. The younger daughter, at age 3.5 years, was diagnosed with DRD caused by THD, and then the diagnosis of the older daughter, at age 11 years, was changed from cerebral palsy to DRD caused by THD.
The diagnosis of dopa-responsive dystonia caused by tyrosine hydroxylase deficiency was determined by whole exome sequencing.
They all treated with low dose levodopa and benserazide tablets.
The boy had a very good therapeutic effect, and he could walk very well by the second day of treatment. The younger sister of the siblings had a partial therapeutic effect, but her elder sister was only little effective with a milder improvement of dystonia and improvement of myodynamia.
The characteristics of THD are heterogeneous, and its phenotypes are classified as type A or type B according to increasing severity. Generally, L-dopa has a good therapeutic effect in cases with type A phenotypes. We reviewed 87 cases of reported in the literature and found that c.698G>A and c.707T>C are hot spot mutations. Changes on cerebral magnetic resonance imaging were nonspecific. Analysis of neurotransmitter levels in cerebrospinal fluid is an invasive means of achieving a biochemical diagnosis.
酪氨酸羟化酶缺乏症(THD)是多巴反应性肌张力障碍(DRD)的一种罕见病因。尽管左旋多巴治疗可改善DRD的症状,但THD发病率低可能导致误诊。因此,医生了解THD作为DRD的病因很重要。
我们报告3例THD病例。一名5岁患DRD的男孩被诊断为THD,发现其TH基因存在复合杂合突变,包括来自母亲的TH:c.647G>C和来自父亲的TH:c.646G>A。两名女性同胞也被发现分别有来自母亲的TH:c.698G>A和来自父亲的TH:c.710T>C。小女儿3.5岁时被诊断为由THD引起的DRD,随后11岁的大女儿的诊断从脑瘫改为由THD引起的DRD。
通过全外显子组测序确定了由酪氨酸羟化酶缺乏引起的多巴反应性肌张力障碍的诊断。
他们均接受低剂量左旋多巴和苄丝肼片治疗。
男孩治疗效果非常好,治疗第二天就能很好地行走。同胞中的妹妹有部分治疗效果,但姐姐效果不佳,肌张力障碍仅稍有改善,肌力有所提高。
THD的特征具有异质性,其表型根据严重程度增加分为A或B型。一般来说,L-多巴对A型表型病例有良好的治疗效果。我们回顾了文献中报道的87例病例,发现c.698G>A和c.707T>C是热点突变。脑磁共振成像的改变不具有特异性。脑脊液神经递质水平分析是实现生化诊断的一种有创手段。