Department of Informatics, I12-Chair of Bioinformatics and Computational Biology, Technical University of Munich (TUM), Boltzmannstrasse 3, 85748, Garching, Munich, Germany.
TUM Graduate School, Center of Doctoral Studies in Informatics and Its Applications (CeDoSIA), 85748, Garching, Germany.
BMC Bioinformatics. 2020 Oct 13;21(1):452. doi: 10.1186/s12859-020-03759-0.
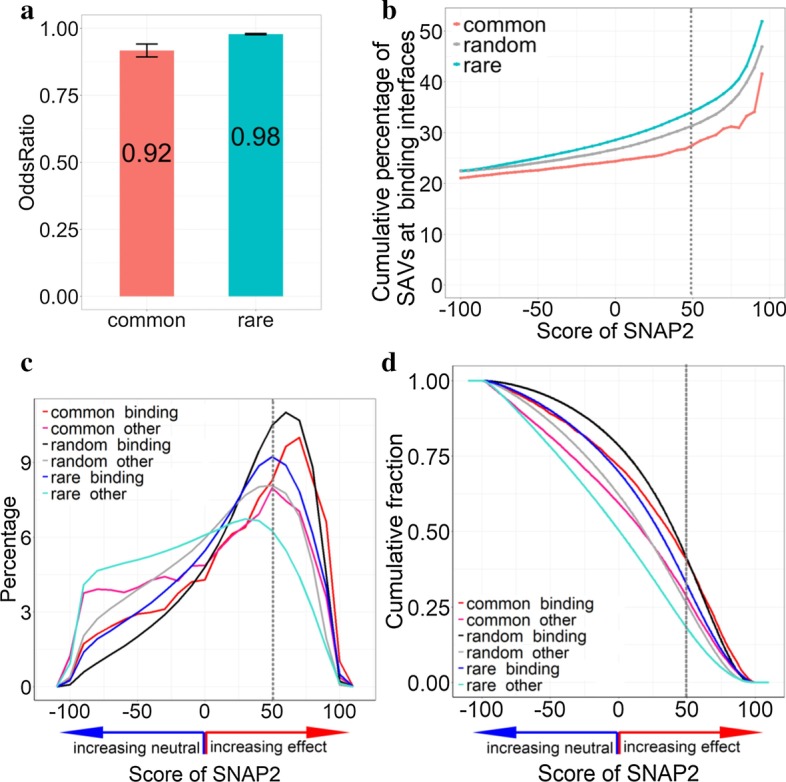
Any two unrelated people differ by about 20,000 missense mutations (also referred to as SAVs: Single Amino acid Variants or missense SNV). Many SAVs have been predicted to strongly affect molecular protein function. Common SAVs (> 5% of population) were predicted to have, on average, more effect on molecular protein function than rare SAVs (< 1% of population). We hypothesized that the prevalence of effect in common over rare SAVs might partially be caused by common SAVs more often occurring at interfaces of proteins with other proteins, DNA, or RNA, thereby creating subgroup-specific phenotypes. We analyzed SAVs from 60,706 people through the lens of two prediction methods, one (SNAP2) predicting the effects of SAVs on molecular protein function, the other (ProNA2020) predicting residues in DNA-, RNA- and protein-binding interfaces.
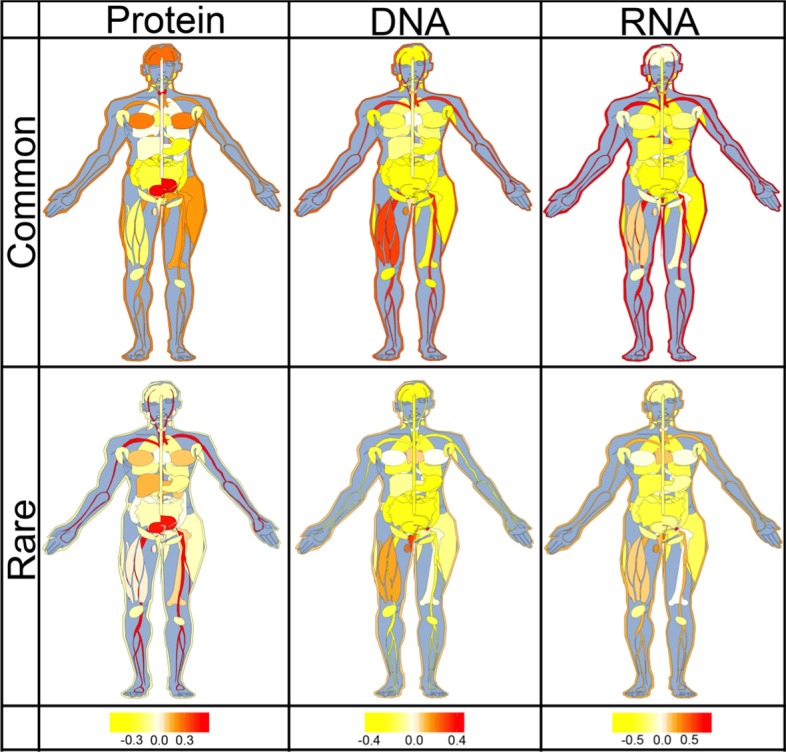
Three results stood out. Firstly, SAVs predicted to occur at binding interfaces were predicted to more likely affect molecular function than those predicted as not binding (p value < 2.2 × 10). Secondly, for SAVs predicted to occur at binding interfaces, common SAVs were predicted more strongly with effect on protein function than rare SAVs (p value < 2.2 × 10). Restriction to SAVs with experimental annotations confirmed all results, although the resulting subsets were too small to establish statistical significance for any result. Thirdly, the fraction of SAVs predicted at binding interfaces differed significantly between tissues, e.g. urinary bladder tissue was found abundant in SAVs predicted at protein-binding interfaces, and reproductive tissues (ovary, testis, vagina, seminal vesicle and endometrium) in SAVs predicted at DNA-binding interfaces.
Overall, the results suggested that residues at protein-, DNA-, and RNA-binding interfaces contributed toward predicting that common SAVs more likely affect molecular function than rare SAVs.
任意两个不相关的人之间存在约 20000 个错义突变(也称为 SAVs:单个氨基酸变异或错义 SNV)。许多 SAV 被预测会强烈影响分子蛋白功能。常见 SAV(>5%的人群)平均比罕见 SAV(<1%的人群)对分子蛋白功能的影响更大。我们假设,常见 SAV 比罕见 SAV 更常见于蛋白质与其他蛋白质、DNA 或 RNA 的界面,从而产生亚组特异性表型,这可能部分导致常见 SAV 比罕见 SAV 的效应更为普遍。我们通过两种预测方法,即一种(SNAP2)预测 SAV 对分子蛋白功能的影响,另一种(ProNA2020)预测 DNA、RNA 和蛋白质结合界面残基的方法,分析了来自 60706 人的 SAV。
有三个结果脱颖而出。首先,预测发生在结合界面的 SAV 比预测不发生结合的 SAV 更有可能影响分子功能(p 值<2.2×10)。其次,对于预测发生在结合界面的 SAV,常见 SAV 比罕见 SAV 更强烈地预测对蛋白质功能有影响(p 值<2.2×10)。限制具有实验注释的 SAV 证实了所有结果,尽管结果子集太小,无法对任何结果建立统计学意义。第三,预测发生在结合界面的 SAV 数量在组织之间存在显著差异,例如,在蛋白质结合界面预测的 SAV 中,膀胱组织丰富,在 DNA 结合界面预测的 SAV 中,生殖组织(卵巢、睾丸、阴道、精囊和子宫内膜)丰富。
总的来说,结果表明,蛋白质、DNA 和 RNA 结合界面的残基有助于预测常见 SAV 比罕见 SAV 更有可能影响分子功能。