Miele Evelina, Di Giannatale Angela, Crocoli Alessandro, Cozza Raffaele, Serra Annalisa, Castellano Aurora, Cacchione Antonella, Cefalo Maria Giuseppina, Alaggio Rita, De Pasquale Maria Debora
Department of Paediatric Haematology/Oncology Cell and Gene Therapy, Bambino Gesù Children's Hospital, IRCCS, Rome, Italy.
Department of Surgery, Bambino Gesù Children's Hospital, IRCCS, Rome, Italy.
Front Oncol. 2020 Oct 15;10:554388. doi: 10.3389/fonc.2020.554388. eCollection 2020.
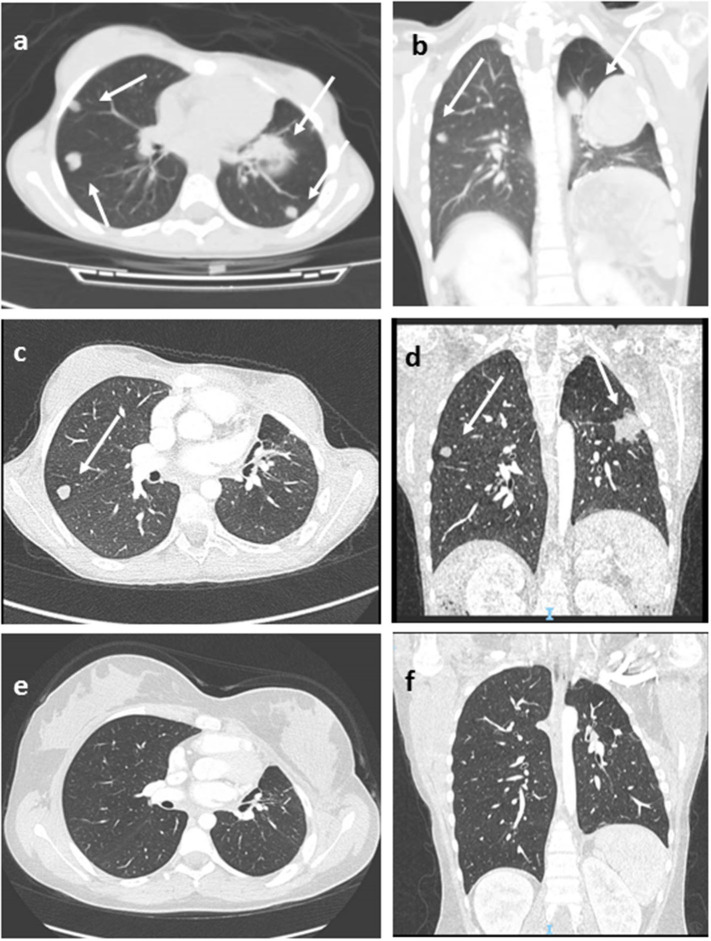
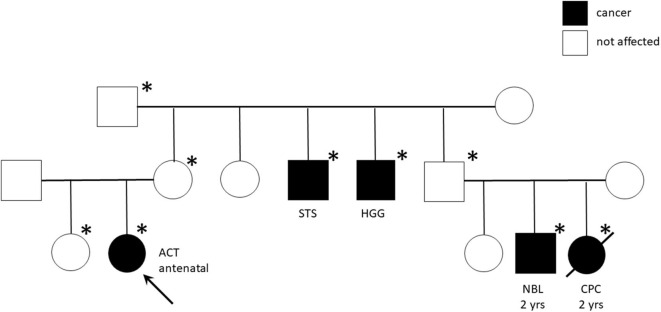
Pediatric adrenocortical tumors (ACTs) are very rare endocrine neoplasms in childhood. In this study, we performed a retrospective analysis of children with ACT treated at our institution by examining clinical and genetic disease features, treatment strategies, and outcomes. We retrospectively analyzed a cohort of 13 children treated at the Bambino Gesù Children's Hospital from November 2010 to March 2020. The median age at diagnosis was 17 months (range = 0-82 months). The female: male ratio was 3.3/1. Mixed symptomatology (>1 hormone abnormality) was the most common presentation (46.1%). In three cases, the tumor was detected during prenatal or perinatal echographic screening. All patients presented with localized disease at diagnosis and underwent total adrenalectomy. Six patients were identified as having malignancies according to the Wieneke scoring system, five benign, and two undetermined. Seven patients underwent mitotane adjuvant therapy for 12 months. There was metastatic disease in three patients, with no correlation with age or Wieneke score. The most common sites of metastases were the liver and lungs. Metastatic patients were treated with surgery ( = 2), mitotane ( = 1), chemotherapy ( = 2) associated with anti-EGFR ( = 1), or immunotherapy with anti-PD1 (pembrolizumab) ( = 1); two patients achieved complete disease remission. Overall 2- and 5-year survival rates were 100%, with a median follow-up of 5 years (range = 2-9.5 years). Two- and 5-year disease free survival was 76.9 and 84.6%, respectively (95% confidence interval = -66.78-114.76 months). All patients are alive, 12 without disease, and one with stable disease. Genetic analyses showed TP53 germline mutations in six of eight patients analyzed (five inherited, one ). One patient had Beckwith-Wiedemann syndrome, with mosaic paternal uniparental disomy of chromosome 11, in both neoplastic and healthy adrenal tissue. We report the cases of 13 patients treated for ACT, including 12 aged <4 years at diagnosis, with a relative short time from symptoms onset. Our cohort experienced an excellent prognosis. TP53 mutation was found in 75% of tested patients (6/8) confirming the need to perform genetic tests and familial counseling in this disease.
儿童肾上腺皮质肿瘤(ACTs)是儿童期非常罕见的内分泌肿瘤。在本研究中,我们通过检查临床和遗传疾病特征、治疗策略及预后,对在我们机构接受治疗的ACT患儿进行了回顾性分析。我们回顾性分析了2010年11月至2020年3月在 Bambino Gesù儿童医院接受治疗的13名儿童队列。诊断时的中位年龄为17个月(范围 = 0 - 82个月)。女性与男性比例为3.3/1。混合症状(>1种激素异常)是最常见的表现(46.1%)。在3例中,肿瘤是在产前或围产期超声筛查时发现的。所有患者诊断时均表现为局限性疾病,并接受了肾上腺全切术。根据Wieneke评分系统,6例患者被确定为恶性肿瘤,5例为良性,2例未确定。7例患者接受了12个月的米托坦辅助治疗。3例患者出现转移,与年龄或Wieneke评分无关。最常见的转移部位是肝脏和肺部。转移患者接受了手术(= 2)、米托坦(= 1)、与抗EGFR联合的化疗(= 2)或抗PD1(帕博利珠单抗)免疫治疗(= 1);2例患者实现了疾病完全缓解。总体2年和5年生存率均为100%,中位随访时间为5年(范围 = 2 - 9.5年)。2年和5年无病生存率分别为76.9%和84.6%(95%置信区间 = -66.78 - 114.76个月)。所有患者均存活,12例无疾病,1例疾病稳定。基因分析显示,在8例分析患者中的6例存在TP53种系突变(5例遗传,1例未知)。1例患者患有贝克威思-维德曼综合征,在肿瘤性和健康肾上腺组织中均存在11号染色体的镶嵌型父源单亲二体。我们报告了13例接受ACT治疗患者的病例,其中12例诊断时年龄<4岁,从症状出现到诊断的时间相对较短。我们的队列预后良好。在75%的检测患者(6/8)中发现了TP53突变,证实了在这种疾病中进行基因检测和家族咨询的必要性。