Department of Microbiology and Molecular Genetics, McGovern Medical School at UTHealth, Houston, TX USA.
MD Anderson UTHealth Graduate School at UTHealth, Houston, TX USA.
Open Biol. 2020 Nov;10(11):200282. doi: 10.1098/rsob.200282. Epub 2020 Nov 25.
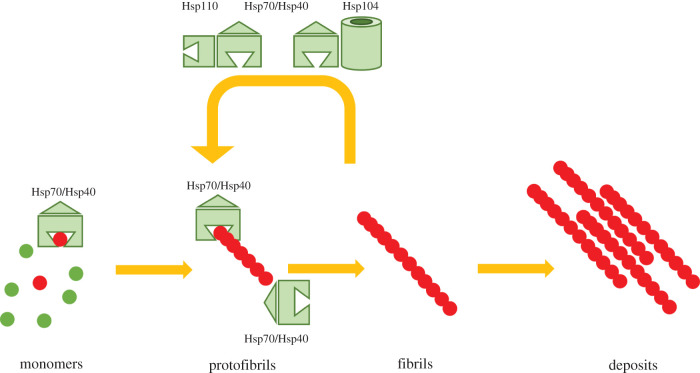
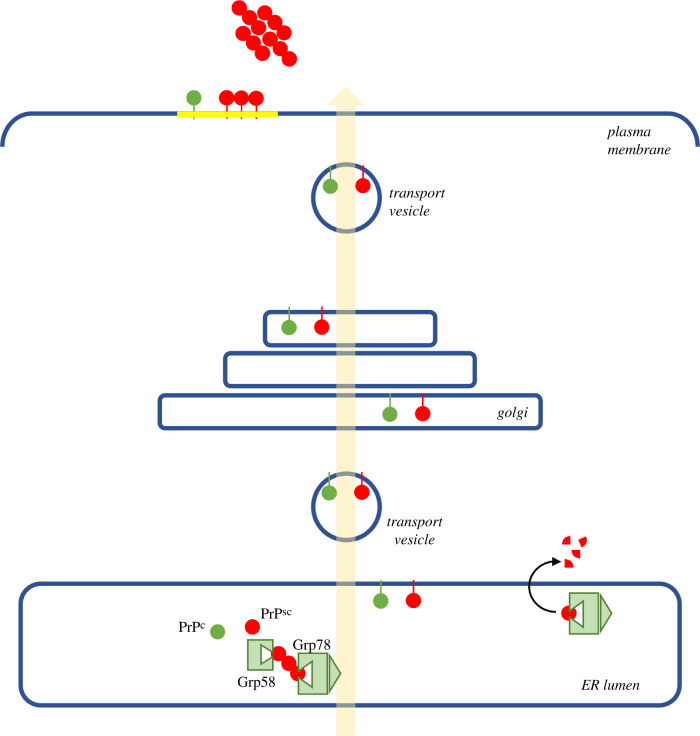
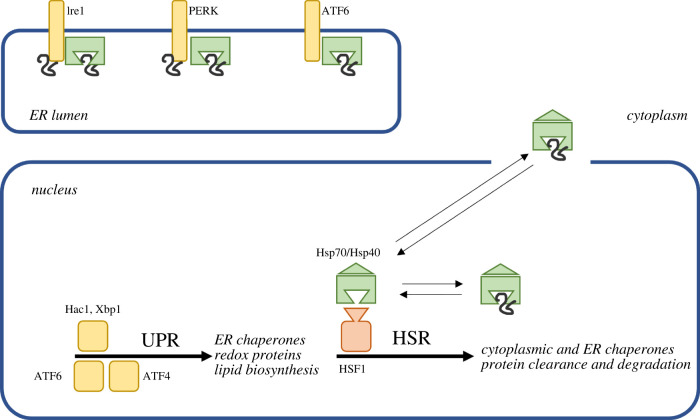
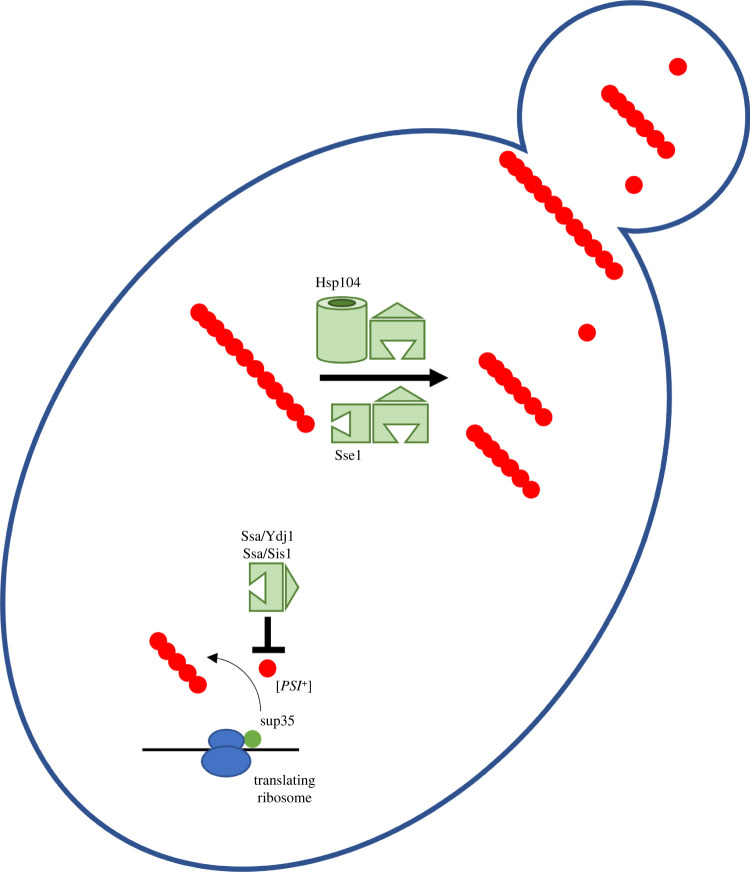
Several neurodegenerative diseases of humans and animals are caused by the misfolded prion protein (PrP), a self-propagating protein infectious agent that aggregates into oligomeric, fibrillar structures and leads to cell death by incompletely understood mechanisms. Work in multiple biological model systems, from simple baker's yeast to transgenic mouse lines, as well as studies, has illuminated molecular and cellular modifiers of prion disease. In this review, we focus on intersections between PrP and the proteostasis network, including unfolded protein stress response pathways and roles played by the powerful regulators of protein folding known as protein chaperones. We close with analysis of promising therapeutic avenues for treatment enabled by these studies.
几种人类和动物的神经退行性疾病是由错误折叠的朊病毒蛋白(PrP)引起的,这种自我传播的蛋白质感染因子会聚集形成寡聚体、纤维状结构,并通过尚未完全了解的机制导致细胞死亡。来自多种生物模型系统的工作,从简单的面包酵母到转基因小鼠系,以及临床研究,都阐明了朊病毒病的分子和细胞修饰物。在这篇综述中,我们重点关注 PrP 与蛋白质稳态网络之间的交叉,包括未折叠蛋白应激反应途径以及被称为蛋白质伴侣的强大折叠蛋白调节剂所发挥的作用。我们以这些研究为基础,分析了有希望的治疗方法。