Institut für Medizinische Genetik und Humangenetik, Charité - Universitätsmedizin Berlin, corporate member of Freie Universität Berlin, Humboldt Universität zu Berlin, and Berlin Institute of Health, Berlin, Germany.
Max Planck Institute for Molecular Genetics, RG Development & Disease, Berlin, Germany.
J Inherit Metab Dis. 2021 Jul;44(4):972-986. doi: 10.1002/jimd.12341. Epub 2021 Feb 4.
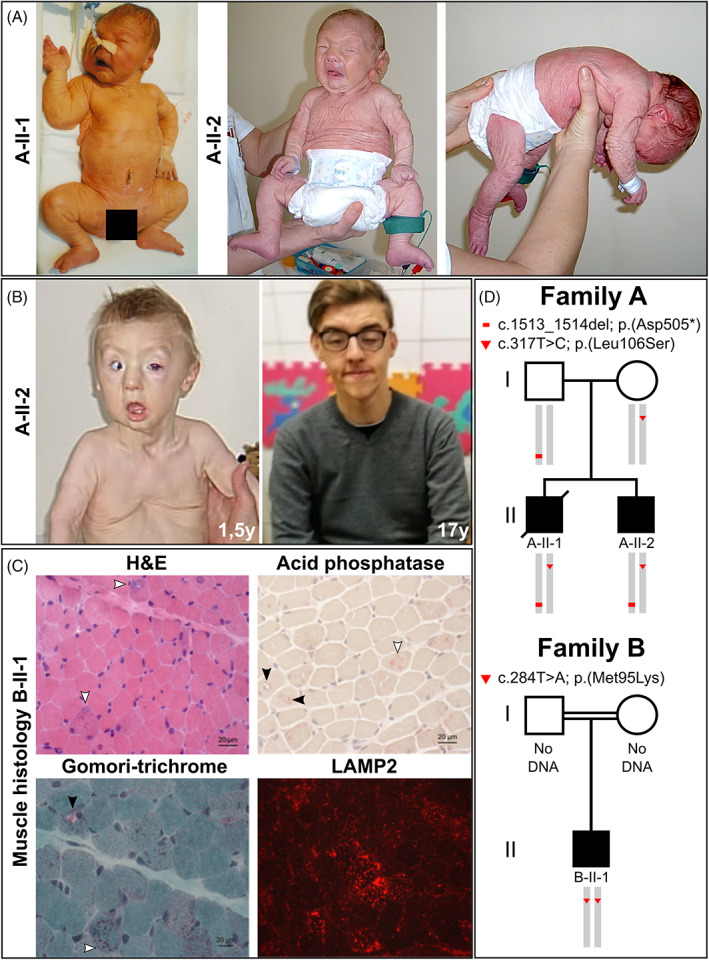
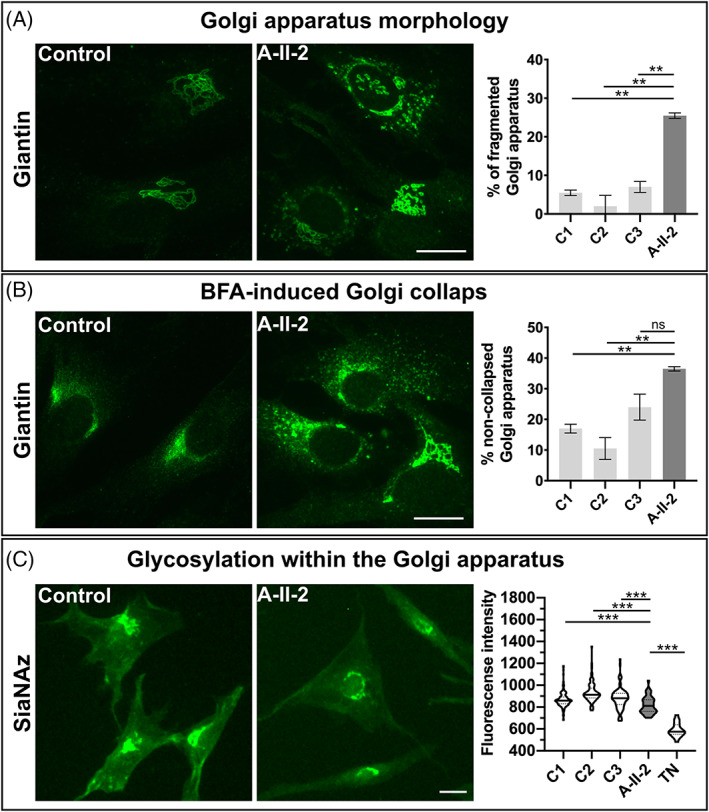
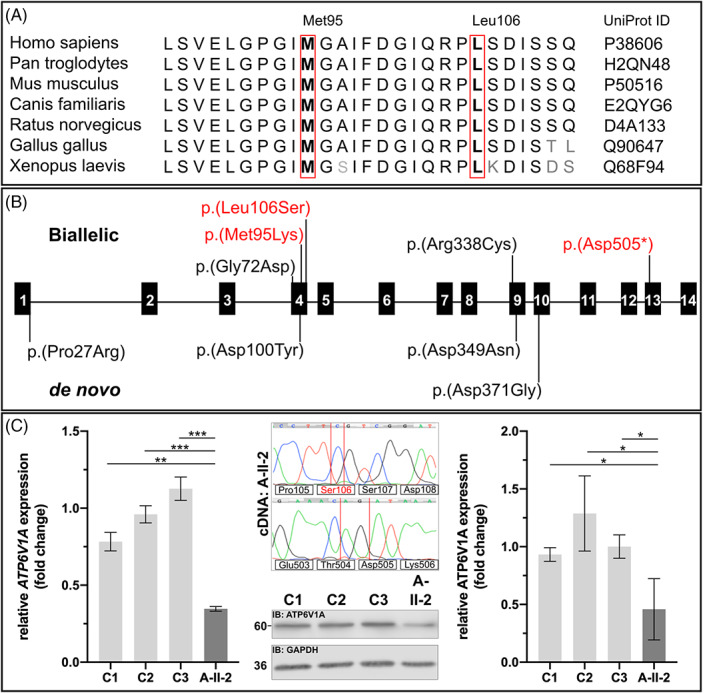
Several inborn errors of metabolism show cutis laxa as a highly recognizable feature. One group of these metabolic cutis laxa conditions is autosomal recessive cutis laxa type 2 caused by defects in v-ATPase components or the mitochondrial proline cycle. Besides cutis laxa, muscular hypotonia and cardiac abnormalities are hallmarks of autosomal recessive cutis laxa type 2D (ARCL2D) due to pathogenic variants in ATP6V1A encoding subunit A of the v-ATPase. Here, we report on three affected individuals from two families with ARCL2D in whom we performed whole exome and Sanger sequencing. We performed functional studies in fibroblasts from one individual, summarized all known probands' clinical, molecular, and biochemical features and compared them, also to other metabolic forms of cutis laxa. We identified novel missense and the first nonsense variant strongly affecting ATP6V1A expression. All six ARCL2D affected individuals show equally severe cutis laxa and dysmorphism at birth. While for one no information was available, two died in infancy and three are now adolescents with mild or absent intellectual disability. Muscular weakness, ptosis, contractures, and elevated muscle enzymes indicated a persistent myopathy. In cellular studies, a fragmented Golgi compartment, a delayed Brefeldin A-induced retrograde transport and glycosylation abnormalities were present in fibroblasts from two individuals. This is the second and confirmatory report on pathogenic variants in ATP6V1A as the cause of this extremely rare condition and the first to describe a nonsense allele. Our data highlight the tremendous clinical variability of ATP6V1A related phenotypes even within the same family.
几种先天性代谢缺陷表现出松弛皮肤症,这是一种高度可识别的特征。这些代谢性松弛皮肤症中的一组是常染色体隐性遗传松弛皮肤症 2 型,由 v-ATPase 成分或线粒体脯氨酸循环的缺陷引起。除了松弛皮肤症外,由于 v-ATPase 编码亚单位 A 的 ATP6V1A 中的致病性变异,常染色体隐性遗传松弛皮肤症 2D(ARCL2D)还具有肌肉张力减退和心脏异常等特征。在这里,我们报告了两个家族中的三个受影响的个体患有 ARCL2D,我们对他们进行了全外显子组和 Sanger 测序。我们对来自一个个体的成纤维细胞进行了功能研究,总结了所有已知先证者的临床、分子和生化特征,并对其进行了比较,也与其他代谢性松弛皮肤症进行了比较。我们发现了新的错义和第一个严重影响 ATP6V1A 表达的无义变异。所有 6 名 ARCL2D 受影响的个体在出生时均表现出同样严重的松弛皮肤症和畸形。虽然有一个没有信息,但是两个在婴儿期死亡,现在有三个是青少年,有轻度或无智力残疾。肌肉无力、上睑下垂、挛缩和肌肉酶升高表明存在持续性肌病。在细胞研究中,来自两个人的成纤维细胞中存在碎片化的高尔基体区室、Brefeldin A 诱导的逆行运输延迟和糖基化异常。这是第二次也是证实性的报道,表明 ATP6V1A 中的致病性变异是这种极其罕见疾病的原因,也是首次描述无义等位基因。我们的数据突出了即使在同一家庭中,ATP6V1A 相关表型的巨大临床变异性。