Amburgey Kimberly, Acker Meryl, Saeed Samia, Amin Reshma, Beggs Alan H, Bönnemann Carsten G, Brudno Michael, Constantinescu Andrei, Dastgir Jahannaz, Diallo Mamadou, Genetti Casie A, Glueck Michael, Hewson Stacy, Hum Courtney, Jain Minal S, Lawlor Michael W, Meyer Oscar H, Nelson Leslie, Sultanum Nicole, Syed Faiza, Tran Tuyen, Wang Ching H, Dowling James J
From the Division of Neurology (K.A.), Genetics and Genome Biology (K.A., M.A., J.J.D., M.B., N.S.), Division of Respiratory Medicine (R.A., F.S., T.T.), Centre for Computational Medicine (M.B., N.S.), Division of Emergency Medicine (M.D.), and Division of Clinical and Metabolic Genetics (S.H.), Hospital for Sick Children; Princess Margaret Hospital (S.S.), Department of Medical Oncology and Hematology; University of Toronto (R.A.), Ontario, Canada; The Manton Center for Orphan Disease Research (A.H.B., C.A.G.), Division of Genetics and Genomics, Boston Children's Hospital, Harvard Medical School, MA; National Institute of Neurological Disorders and Stroke (C.G.B.), Neuromuscular and Neurogenetic Disorders of Childhood Section, and Clinical Research Center (M.S.J.), Rehabilitation Medicine Department, NIH, Bethesda, MD; Department of Computer Science (M.B., M.G., N.S.), University of Toronto, Ontario, Canada; Columbia University Irving Medical Center (A.C.), Division of Pediatric Pulmonology, New York, NY; Goryeb Children's Hospital (J.D.), Department of Pediatric Neurology, Morristown, NJ; Mount Sinai Hospital (C.H.), Prenatal Diagnosis and Medical Genetics, Toronto, Ontario, Canada; Medical College of Wisconsin (M.W.L.), Department of Pathology and Laboratory Medicine, Milwaukee; Children's Hospital of Philadelphia (O.H.M.), Division of Pulmonology, PA; UT Southwestern Medical Center (L.N.), Department of Physical Therapy, Dallas, TX; and Driscoll Children's Hospital (C.H.W.), Division of Neurology, Texas A&M University, Corpus Christi.
Neurology. 2021 Mar 9;96(10):e1425-e1436. doi: 10.1212/WNL.0000000000011458. Epub 2021 Jan 4.
Nemaline myopathy (NM) is a rare neuromuscular condition with clinical and genetic heterogeneity. To establish disease natural history, we performed a cross-sectional study of NM, complemented by longitudinal assessment and exploration of pilot outcome measures.
Fifty-seven individuals with NM were recruited at 2 family workshops, including 16 examined at both time points. Participants were evaluated by clinical history and physical examination. Functional outcome measures included the Motor Function Measure (MFM), pulmonary function tests (PFTs), myometry, goniometry, and bulbar assessments.
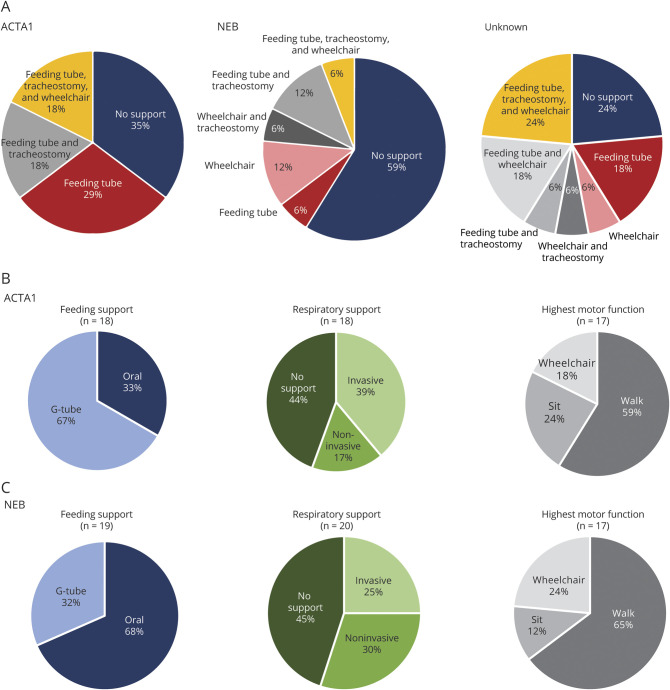
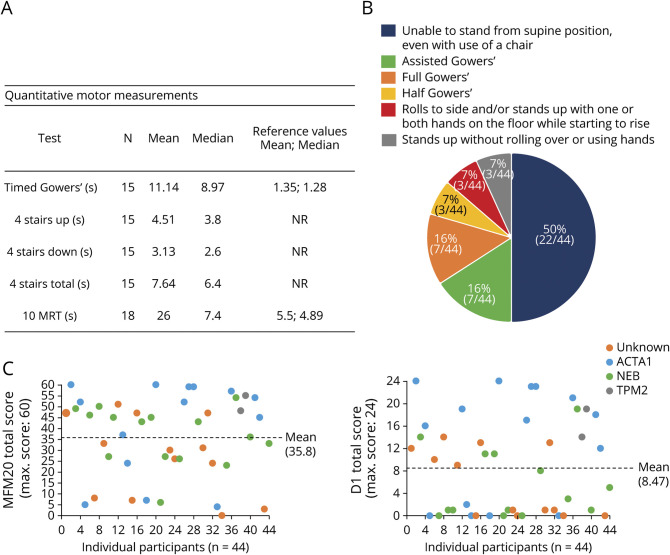
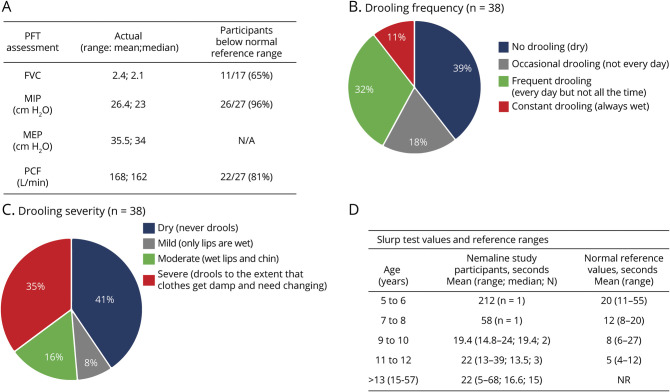
The most common clinical classification was typical congenital (54%), whereas 42% had more severe presentations. Fifty-eight percent of individuals needed mechanical support, with 26% requiring wheelchair, tracheostomy, and feeding tube. The MFM scale was performed in 44 of 57 participants and showed reduced scores in most with little floor/ceiling effect. Of the 27 individuals completing PFTs, abnormal values were observed in 65%. Last, bulbar function was abnormal in all patients examined, as determined with a novel outcome measure. Genotypes included mutations in (18), (20), and (2). Seventeen individuals were genetically unresolved. Patients with pathogenic and variants were largely similar in clinical phenotype. Patients without genetic resolution had more severe disease.
We present a comprehensive cross-sectional study of NM. Our data identify significant disabilities and support a relatively stable disease course. We identify a need for further diagnostic investigation for the genetically unresolved group. MFM, PFTs, and the slurp test were identified as promising outcome measures for future clinical trials.
杆状体肌病(NM)是一种罕见的神经肌肉疾病,具有临床和遗传异质性。为了确定疾病的自然史,我们对NM进行了一项横断面研究,并辅以纵向评估和探索初步的结局指标。
在2个家庭研讨会上招募了57名NM患者,其中16名在两个时间点均接受了检查。通过临床病史和体格检查对参与者进行评估。功能结局指标包括运动功能测量(MFM)、肺功能测试(PFT)、肌测力测量、关节角度测量和延髓评估。
最常见的临床分类是典型先天性(54%),而42%的患者表现更为严重。58%的个体需要机械支持,其中26%需要轮椅、气管造口术和饲管。57名参与者中有44名进行了MFM量表评估,大多数人的得分降低,几乎没有地板效应/天花板效应。在完成PFT的27名个体中,65%观察到异常值。最后,根据一项新的结局指标测定,所有接受检查的患者延髓功能均异常。基因型包括 (18例)、 (20例)和 (2例)的突变。17名个体的基因未得到明确诊断。携带致病性 和 变异的患者临床表型基本相似。基因未明确诊断的患者病情更严重。
我们对NM进行了一项全面的横断面研究。我们的数据确定了严重的残疾情况,并支持疾病进程相对稳定。我们发现有必要对基因未明确诊断的组进行进一步的诊断研究。MFM、PFT和吞咽试验被确定为未来临床试验中有前景的结局指标。