Department of Biochemistry, Jomo Kenyatta University of Agriculture and Technology, P.O. Box 62000, Nairobi, City Square, Kenya.
Department of Biochemistry and Molecular Biology, University of Buea, P.O. Box 63, Buea, South West Region, Cameroon.
Sci Rep. 2021 Jan 13;11(1):1039. doi: 10.1038/s41598-020-79124-1.
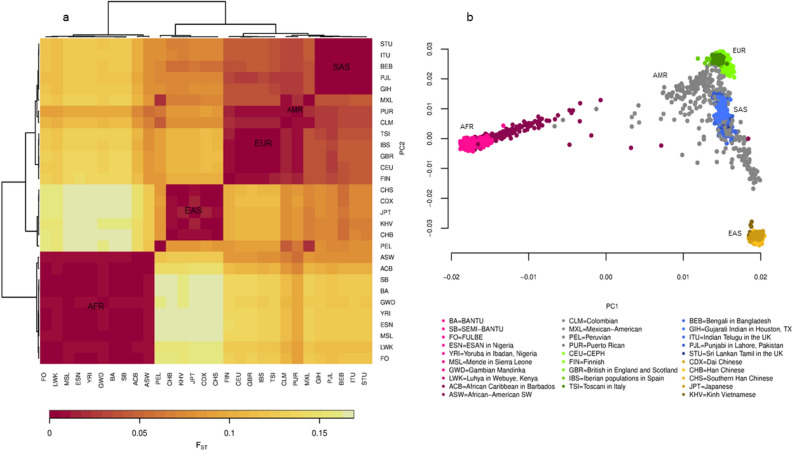
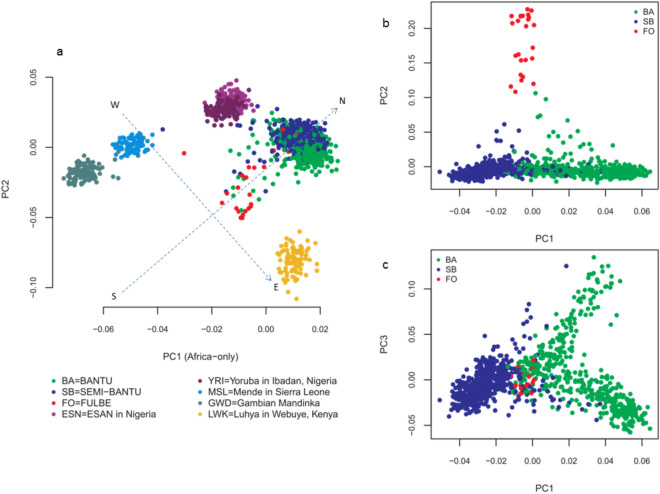
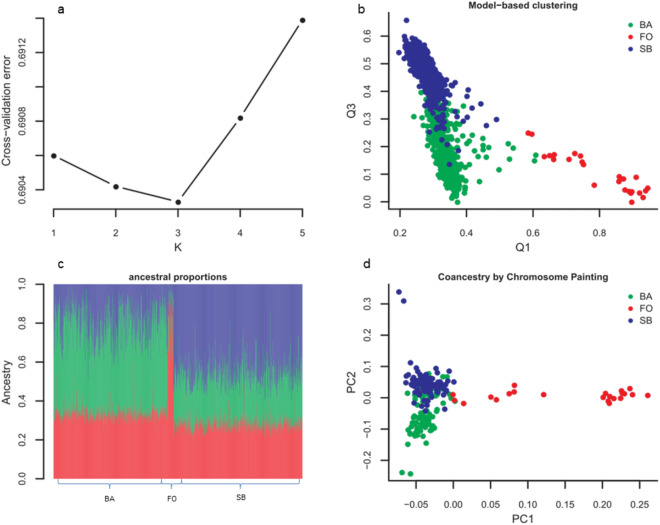
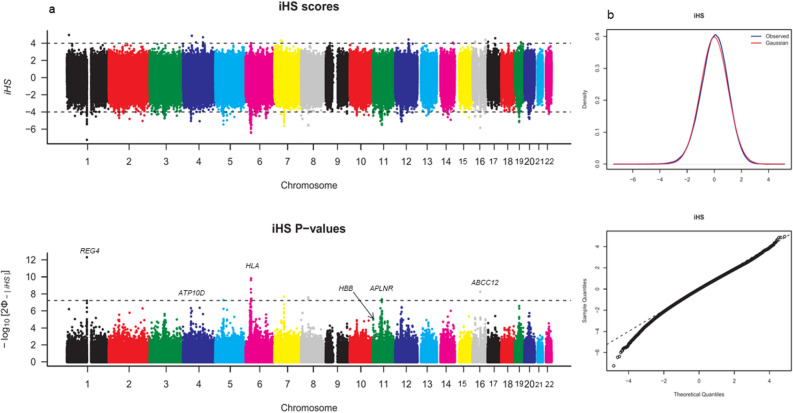
Inferences from genetic association studies rely largely on the definition and description of the underlying populations that highlight their genetic similarities and differences. The clustering of human populations into subgroups (population structure) can significantly confound disease associations. This study investigated the fine-scale genetic structure within Cameroon that may underlie disparities observed with Cameroonian ethnicities in malaria genome-wide association studies in sub-Saharan Africa. Genotype data of 1073 individuals from three regions and three ethnic groups in Cameroon were analyzed using measures of genetic proximity to ascertain fine-scale genetic structure. Model-based clustering revealed distinct ancestral proportions among the Bantu, Semi-Bantu and Foulbe ethnic groups, while haplotype-based coancestry estimation revealed possible longstanding and ongoing sympatric differentiation among individuals of the Foulbe ethnic group, and their Bantu and Semi-Bantu counterparts. A genome scan found strong selection signatures in the HLA gene region, confirming longstanding knowledge of natural selection on this genomic region in African populations following immense disease pressure. Signatures of selection were also observed in the HBB gene cluster, a genomic region known to be under strong balancing selection in sub-Saharan Africa due to its co-evolution with malaria. This study further supports the role of evolution in shaping genomes of Cameroonian populations and reveals fine-scale hierarchical structure among and within Cameroonian ethnicities that may impact genetic association studies in the country.
遗传关联研究的推论在很大程度上依赖于对突出其遗传相似性和差异性的基础人群的定义和描述。人群聚类成亚群(群体结构)会严重干扰疾病关联。本研究调查了喀麦隆内部的精细遗传结构,这些结构可能是导致在撒哈拉以南非洲进行的疟疾全基因组关联研究中喀麦隆族群之间存在差异的原因。使用遗传邻近度衡量标准分析了来自喀麦隆三个地区和三个族群的 1073 个人的基因型数据,以确定精细遗传结构。基于模型的聚类揭示了班图、半班图和富尔贝三个族群之间存在明显的祖先比例,而基于单倍型的亲缘关系估计则揭示了富尔贝族群内部以及与他们的班图和半班图族群之间可能存在长期的和正在进行的同域分化。基因组扫描在 HLA 基因区域发现了强烈的选择信号,证实了在巨大疾病压力下,非洲人群中该基因组区域的自然选择是长期存在的。在 HBB 基因簇中也观察到了选择信号,这是一个由于与疟疾共同进化而在撒哈拉以南非洲地区受到强烈平衡选择的基因组区域。本研究进一步支持了进化在塑造喀麦隆人群基因组中的作用,并揭示了喀麦隆族群内部和之间的精细层次结构,这可能会影响该国的遗传关联研究。