Department of Hepatobiliary Surgery, The Second Affiliated Hospital of Zhejiang University School of Medicine, Hangzhou, China.
Department of General Surgery, Hangzhou Mingzhou Hospital, Hangzhou, China.
J Int Med Res. 2021 Feb;49(2):300060520980646. doi: 10.1177/0300060520980646.
Hepatocellular carcinoma (HCC) is a highly malignant tumor with a particularly poor prognosis. The tumor microenvironment (TME) is closely associated with tumorigenesis, progression, and treatment. However, the relationship between TME genes and HCC patient prognosis is poorly understood.
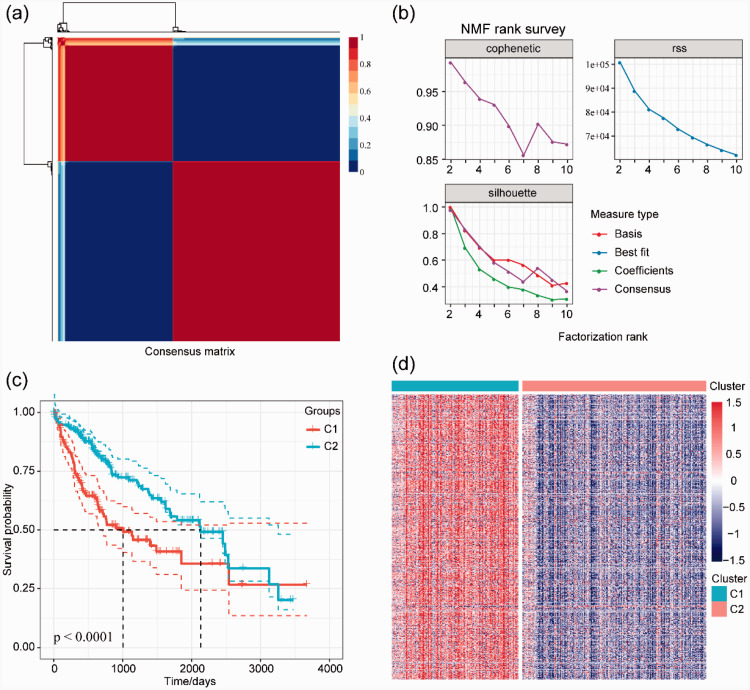
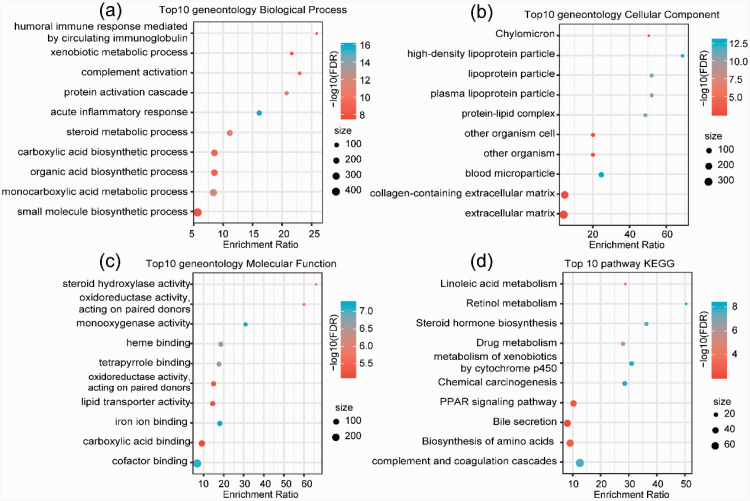
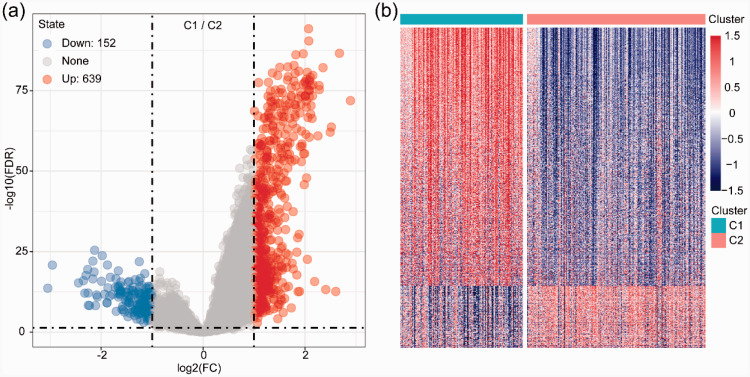
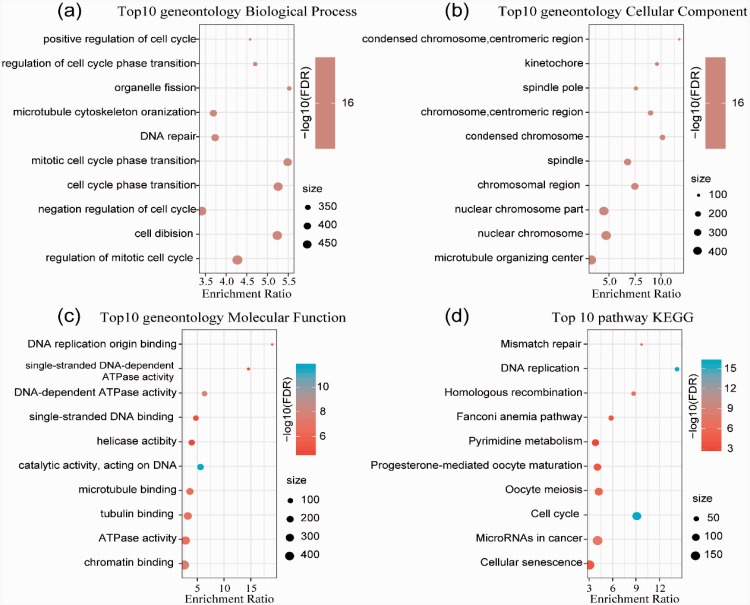
In this study, we identified two prognostic subtypes based on the TME using data from The Cancer Genome Atlas and Gene Expression Omnibus. The Microenvironment Cell Populations-counter method was used to evaluate immune cell infiltration in HCC. Differentially expressed genes between molecular subtypes were calculated with the Limma package, and clusterProfiler was used for Gene Ontology and Kyoto Encyclopedia of Genes and Genomes functional enrichment analyses to identify genes related to the independent subtypes. We also integrated mRNA expression data into our bioinformatics analysis.
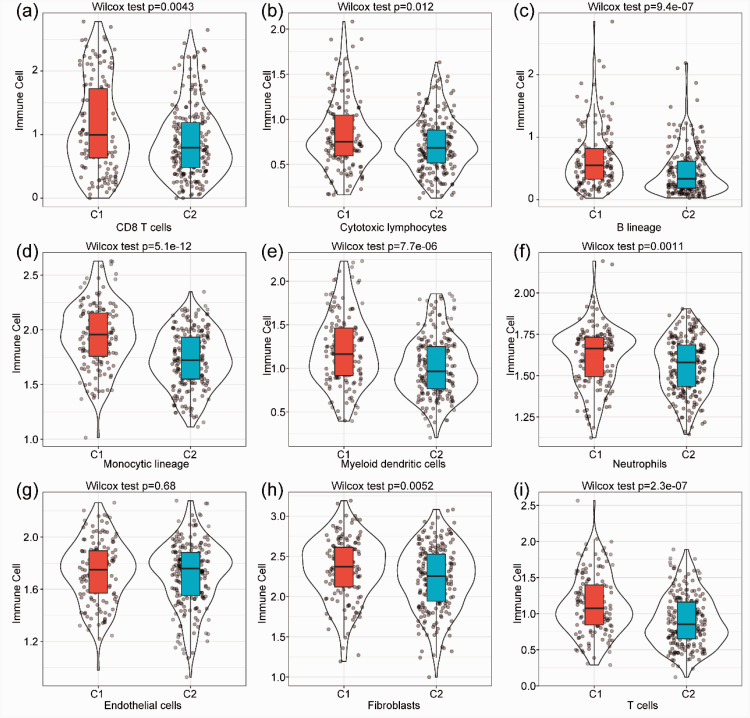
We identified 4227 TME-associated genes and 640 genes related to the prognosis of HCC. We defined two major subtypes (Clusters 1 and 2) based on the analysis of TME-associated gene expression. Cluster 1 was characterized by increased expression of immune-associated genes and a worse prognosis than Cluster 2.
The identification of these HCC subtypes based on the TME provides further insight into the molecular mechanisms and prediction of HCC prognosis.
肝细胞癌(HCC)是一种高度恶性肿瘤,预后尤其差。肿瘤微环境(TME)与肿瘤发生、进展和治疗密切相关。然而,TME 基因与 HCC 患者预后的关系尚不清楚。
本研究基于癌症基因组图谱和基因表达综合数据库的数据,采用微环境细胞群体计数法评估 HCC 中的免疫细胞浸润。使用 Limma 包计算分子亚型之间差异表达的基因,并使用 clusterProfiler 进行基因本体论和京都基因与基因组百科全书功能富集分析,以鉴定与独立亚型相关的基因。我们还将 mRNA 表达数据整合到我们的生物信息学分析中。
我们确定了 4227 个与 TME 相关的基因和 640 个与 HCC 预后相关的基因。我们根据 TME 相关基因表达的分析定义了两个主要亚型(簇 1 和簇 2)。簇 1 的特点是免疫相关基因表达增加,预后比簇 2差。
基于 TME 识别这些 HCC 亚型为 HCC 的分子机制和预后预测提供了更深入的认识。