Department of Genetics and Evolution, Center for Biological Sciences and Health, Federal University of São Carlos (UFSCar), São Carlos, SP, 13565-905, Brazil.
Department of Parasitology, ICB, University of São Paulo (USP), São Paulo, SP, Brazil.
Sci Rep. 2021 Feb 15;11(1):3791. doi: 10.1038/s41598-021-81926-w.
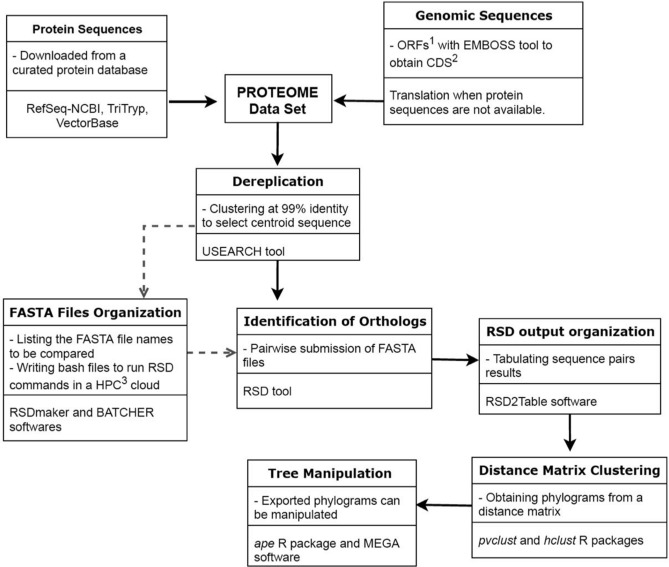
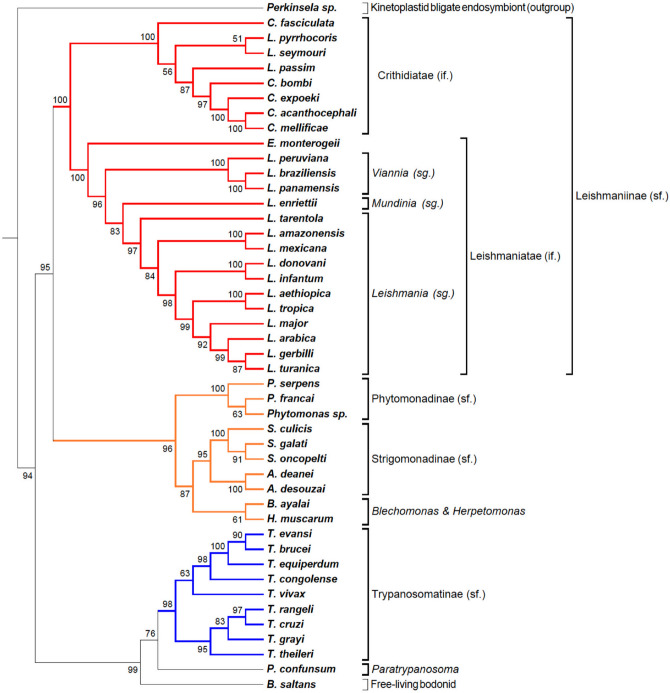
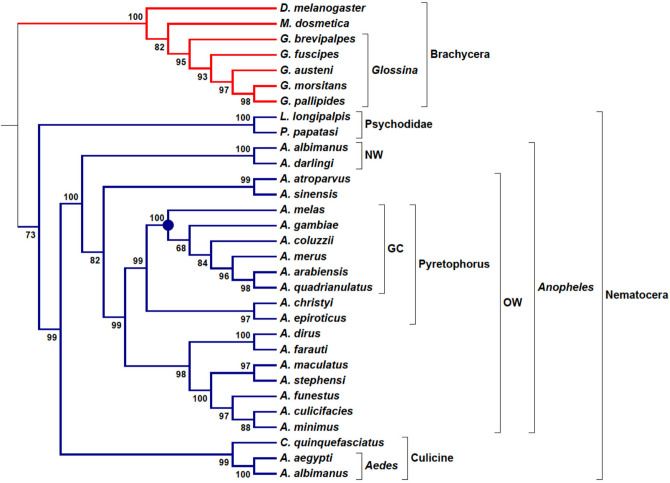
The increasing number of available genomic data allowed the development of phylogenomic analytical tools. Current methods compile information from single gene phylogenies, whether based on topologies or multiple sequence alignments. Generally, phylogenomic analyses elect gene families or genomic regions to construct phylogenomic trees. Here, we presented an alternative approach for Phylogenomics, named TOMM (Total Ortholog Median Matrix), to construct a representative phylogram composed by amino acid distance measures of all pairwise ortholog protein sequence pairs from desired species inside a group of organisms. The procedure is divided two main steps, (1) ortholog detection and (2) creation of a matrix with the median amino acid distance measures of all pairwise orthologous sequences. We tested this approach within three different group of organisms: Kinetoplastida protozoa, hematophagous Diptera vectors and Primates. Our approach was robust and efficacious to reconstruct the phylogenetic relationships for the three groups. Moreover, novel branch topologies could be achieved, providing insights about some phylogenetic relationships between some taxa.
越来越多的基因组数据使得系统发生基因组分析工具得以发展。目前的方法从单基因系统发生信息中编译,无论是基于拓扑结构还是多重序列比对。一般来说,系统发生基因组分析选择基因家族或基因组区域来构建系统发生树。在这里,我们提出了一种用于系统发生基因组学的替代方法,命名为 TOMM(总直系同源中位数矩阵),以构建由一组生物体内所需物种的所有成对直系同源蛋白序列对的氨基酸距离度量组成的代表系统发生图。该过程分为两个主要步骤:(1)直系同源检测和(2)创建具有所有成对直系同源序列中位数氨基酸距离度量的矩阵。我们在三种不同的生物群中测试了这种方法:动基体目原生动物、吸血双翅目媒介和灵长类动物。我们的方法对于重建这三个群体的系统发育关系是稳健和有效的。此外,还可以获得新的分支拓扑结构,提供关于一些分类单元之间的系统发育关系的见解。