Pediatric Research, Medicine Department, University of Oviedo, Oviedo, Spain.
AGC Pediatría, Hospital Universitario Central de Asturias, Oviedo, Spain.
Orphanet J Rare Dis. 2021 Feb 27;16(1):104. doi: 10.1186/s13023-021-01729-0.
X-linked hypophosphatemia (XLH) is a hereditary rare disease caused by loss-of-function mutations in PHEX gene leading tohypophosphatemia and high renal loss of phosphate. Rickets and growth retardation are the major manifestations of XLH in children, but there is a broad phenotypic variability. Few publications have reported large series of patients. Current data on the clinical spectrum of the disease, the correlation with the underlying gene mutations, and the long-term outcome of patients on conventional treatment are needed, particularly because of the recent availability of new specific medications to treat XLH.
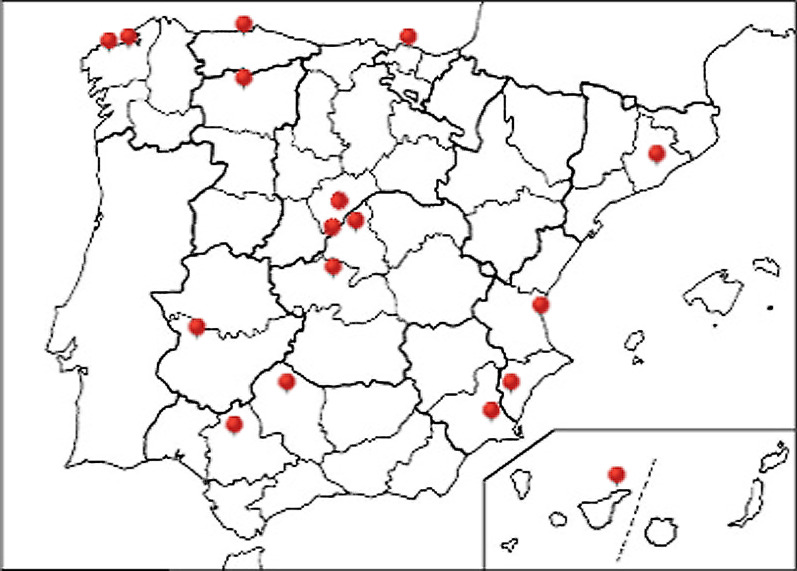
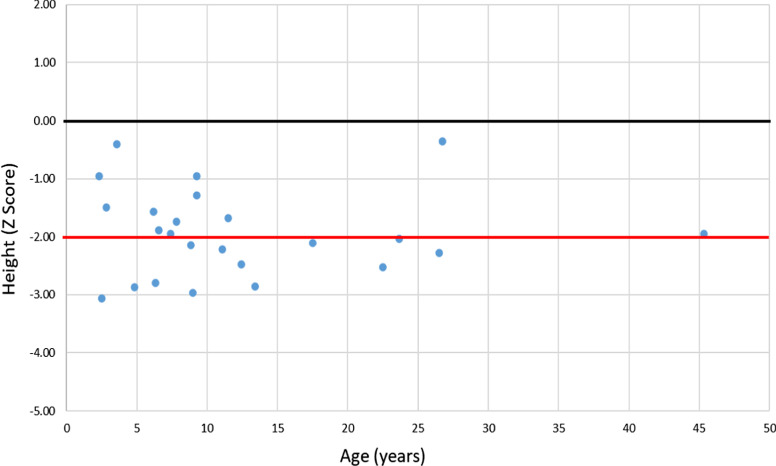
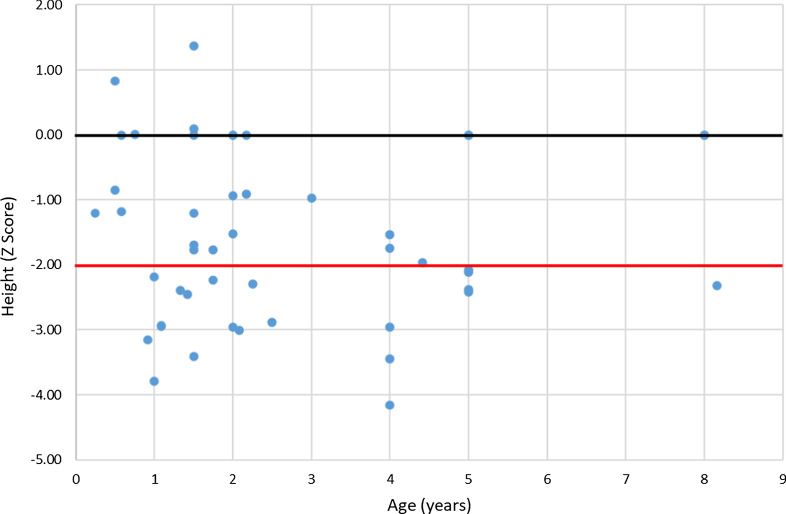
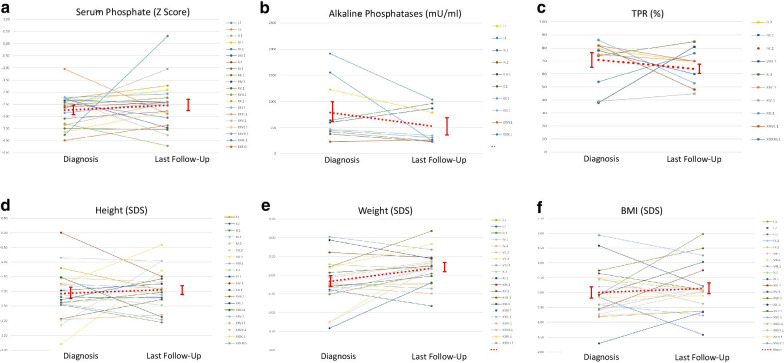
The RenalTube database was used to retrospectively analyze 48 Spanish patients (15 men) from 39 different families, ranging from 3 months to 8 years and 2 months of age at the time of diagnosis (median age of 2.0 years), and with XLH confirmed by genetic analysis. Bone deformities, radiological signs of active rickets and growth retardation were the most common findings at diagnosis. Mean (± SEM) height was - 1.89 ± 0.19 SDS and 55% (22/40) of patients had height SDS below-2. All cases had hypophosphatemia, serum phosphate being - 2.81 ± 0.11 SDS. Clinical manifestations and severity of the disease were similar in both genders. No genotype-phenotype correlation was found. Conventional treatment did not attenuate growth retardation after a median follow up of 7.42 years (IQR = 11.26; n = 26 patients) and failed to normalize serum concentrations of phosphate. Eleven patients had mild hyperparathyroidism and 8 patients nephrocalcinosis.
This study shows that growth retardation and rickets were the most prevalent clinical manifestations at diagnosis in a large series of Spanish pediatric patients with XLH confirmed by mutations in the PHEX gene. Traditional treatment with phosphate and vitamin D supplements did not improve height or corrected hypophosphatemia and was associated with a risk of hyperparathyroidism and nephrocalcinosis. The severity of the disease was similar in males and females.
X 连锁低磷血症(XLH)是一种遗传性罕见疾病,由 PHEX 基因突变导致,导致低磷血症和磷酸盐肾大量丢失。佝偻病和生长迟缓是 XLH 在儿童中的主要表现,但存在广泛的表型变异性。很少有文献报道大量患者的情况。目前需要了解疾病的临床谱、与潜在基因突变的相关性以及患者接受常规治疗的长期结果,特别是因为最近有了治疗 XLH 的新的特定药物。
使用 RenalTube 数据库回顾性分析了 48 名来自 39 个不同家庭的西班牙患者(男 15 名),诊断时年龄为 3 个月至 8 岁零 2 个月(中位数年龄为 2.0 岁),并通过基因分析确认 XLH。骨畸形、活动性佝偻病的放射学征象和生长迟缓是诊断时最常见的发现。平均(±SEM)身高为-1.89±0.19 SDS,40 名患者中有 55%(22/40)的身高 SDS 低于-2。所有病例均有低磷血症,血清磷酸盐为-2.81±0.11 SDS。两性的临床表现和疾病严重程度相似。未发现基因型-表型相关性。经过中位数为 7.42 年(IQR=11.26;n=26 名患者)的常规治疗后,生长迟缓并未减轻,且未能使血清磷酸盐浓度正常化。11 名患者有轻度甲状旁腺功能亢进,8 名患者有肾钙质沉着症。
本研究表明,在经 PHEX 基因突变证实的 XLH 西班牙儿科患者的大型系列中,生长迟缓是最常见的临床表现,佝偻病是最常见的临床表现。用磷酸盐和维生素 D 补充剂进行传统治疗并不能改善身高或纠正低磷血症,且与甲状旁腺功能亢进和肾钙质沉着症的风险相关。疾病的严重程度在男女之间相似。