Xiong Chongxiang, Deng Jin, Wang Xin, Shao Xiaofei, Zhou Qin, Zou Hequn, Zhuang Shougang
Department of Nephrology, The Third Affiliated Hospital of Southern Medical University, Guangzhou, China.
Department of Nephrology, Shanghai East Hospital, Tongji University School of Medicine, Shanghai, China.
Front Pharmacol. 2021 Feb 16;12:636154. doi: 10.3389/fphar.2021.636154. eCollection 2021.
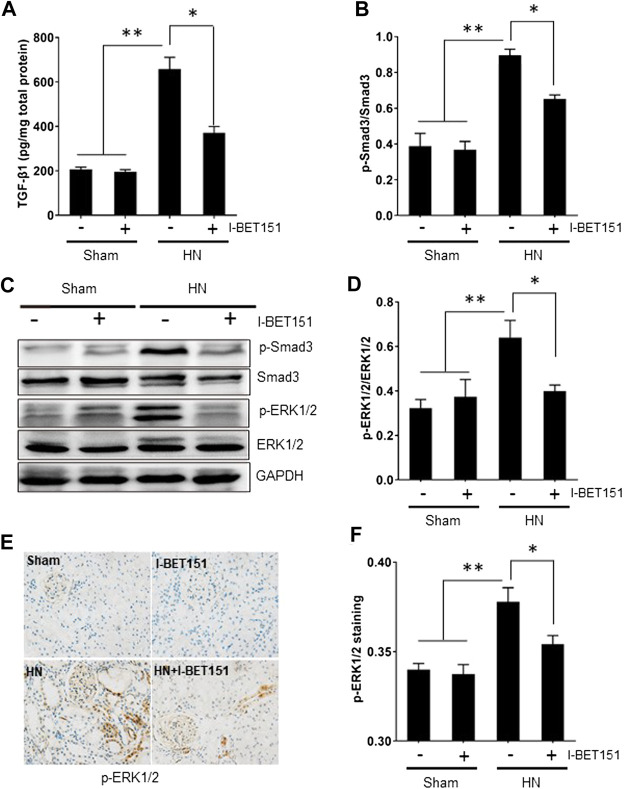
Hyperuricemia is an independent risk factor for renal damage and promotes the progression of chronic kidney disease. In this study, we investigated the effect of I-BET151, a small-molecule inhibitor targeting the bromodomain and extraterminal (BET) proteins, on the development of hyperuricemic nephropathy (HN), and the mechanisms involved. Expression levels of bromodomain-containing protein 2 and 4, but not 3 were increased in the kidney of rats with HN; administration of I-BET151 effectively prevented renal dysfunction, decreased urine microalbumin, and attenuated renal fibrosis as indicated by reduced activation of renal interstitial fibroblasts and expression of fibronectin and collagen I in HN rats. Mechanistic studies show that I-BET151 treatment inhibited transition of renal epithelial cells to a mesenchymal cell type as evidenced by preservation of E-cadherin and reduction of vimentin expression. This was coincident with reduced expression of TGF-β1 and dephosphorylation of Smad3 and ERK1/2. I-BET151 was also effective in inhibiting phosphorylation of NF-κB, expression of multiple cytokines and chemokines, and infiltration of macrophages to the injured kidney. Although there were increased serum levels of uric acid and xanthine oxidase, an enzyme that catalyzes production of uric acid, and decreased expression of renal organic anion transporter 1 and 3 that promote urate excretion in the model of HN, and reduced expression levels of urine uric acid, I-BET151 treatment did not affect these responses. Collectively, our results indicate that I-BET151 alleviates HN by inhibiting epithelial to mesenchymal transition and inflammation in association with blockade of TGF-β, ERK1/2 and NF-κB signaling.
高尿酸血症是肾损伤的独立危险因素,并促进慢性肾脏病的进展。在本研究中,我们研究了靶向溴结构域和额外末端(BET)蛋白的小分子抑制剂I-BET151对高尿酸血症肾病(HN)发展的影响及其相关机制。在HN大鼠的肾脏中,含溴结构域蛋白2和4的表达水平升高,但含溴结构域蛋白3未升高;给予I-BET151可有效预防肾功能障碍,降低尿微量白蛋白,并减轻肾纤维化,这表现为HN大鼠肾间质成纤维细胞的活化减少以及纤连蛋白和I型胶原的表达降低。机制研究表明,I-BET151处理可抑制肾上皮细胞向间充质细胞类型的转变,这表现为E-钙黏蛋白的保留和波形蛋白表达的降低。这与TGF-β1表达的降低以及Smad3和ERK1/2的去磷酸化相一致。I-BET151在抑制NF-κB的磷酸化、多种细胞因子和趋化因子的表达以及巨噬细胞向受损肾脏的浸润方面也有效。尽管在HN模型中血清尿酸水平和催化尿酸生成的酶黄嘌呤氧化酶升高,促进尿酸排泄的肾有机阴离子转运蛋白1和3的表达降低,尿尿酸表达水平降低,但I-BET151处理并未影响这些反应。总体而言,我们的结果表明,I-BET151通过抑制上皮-间充质转化和炎症以及阻断TGF-β、ERK1/2和NF-κB信号传导来减轻HN。