Chemical Biology Laboratory, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Frederick, MD 21702, USA.
Department of Biology and Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA.
Org Biomol Chem. 2021 Sep 22;19(36):7843-7854. doi: 10.1039/d1ob01120k.
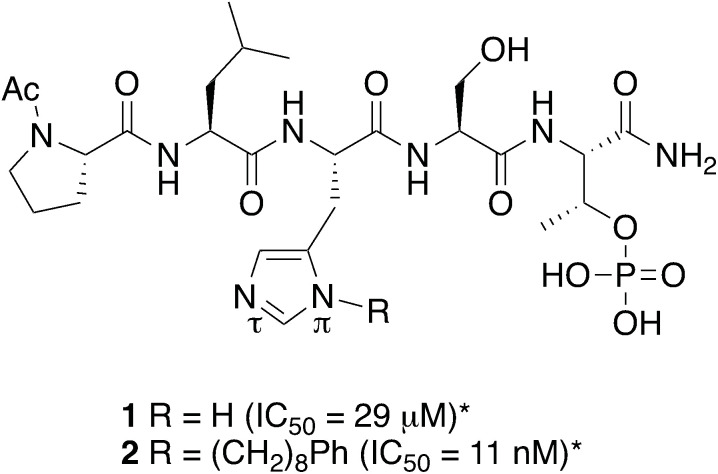
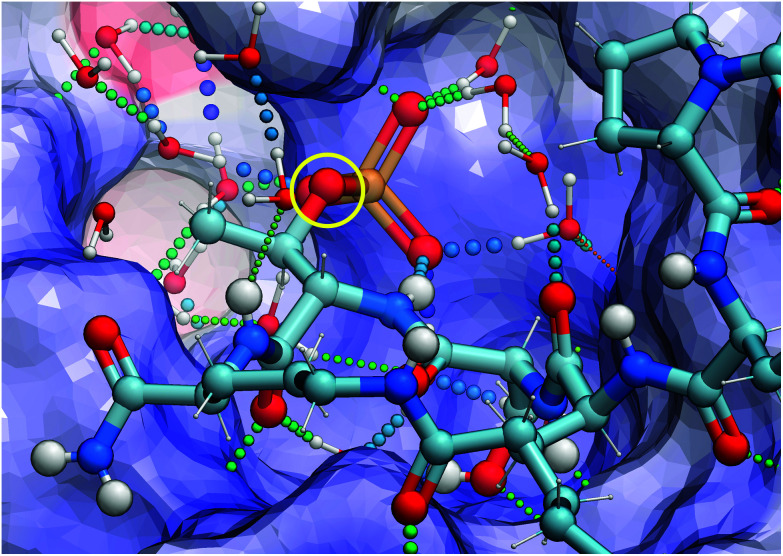
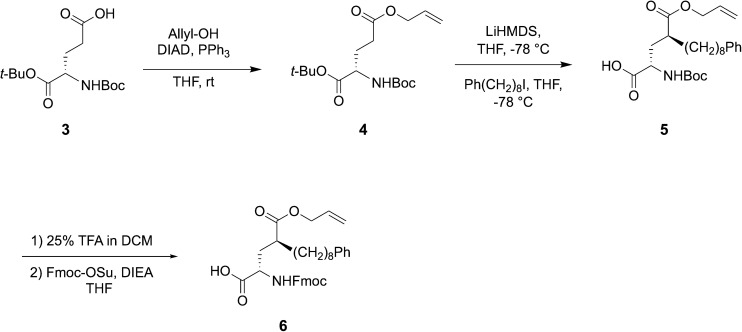
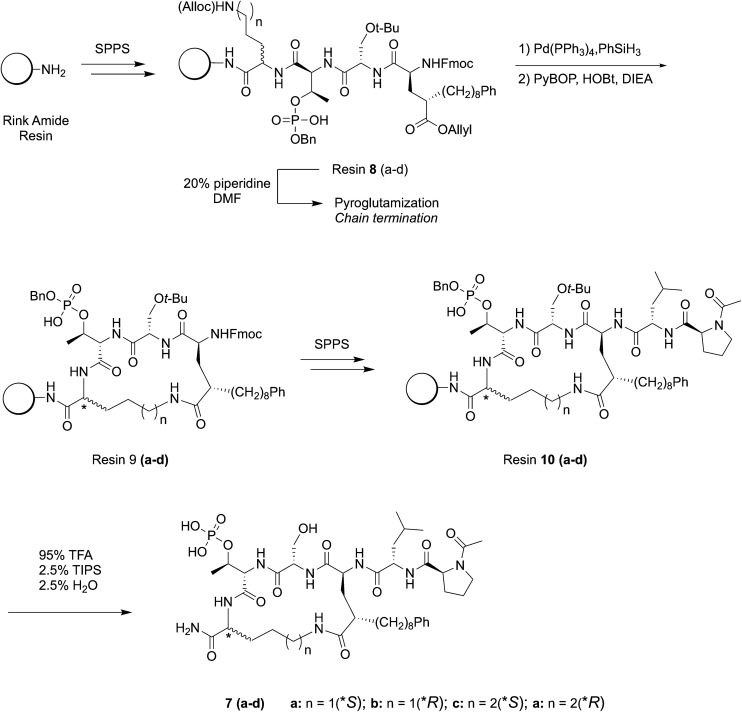
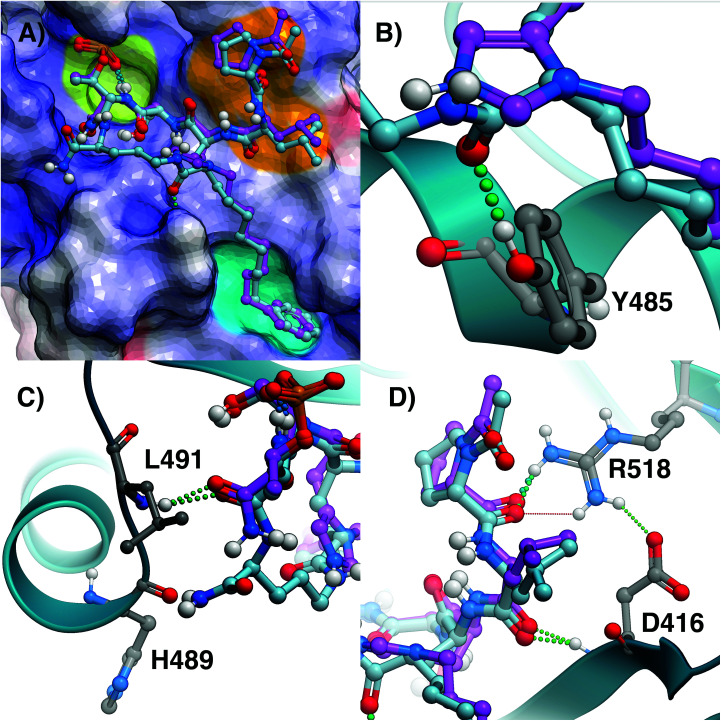
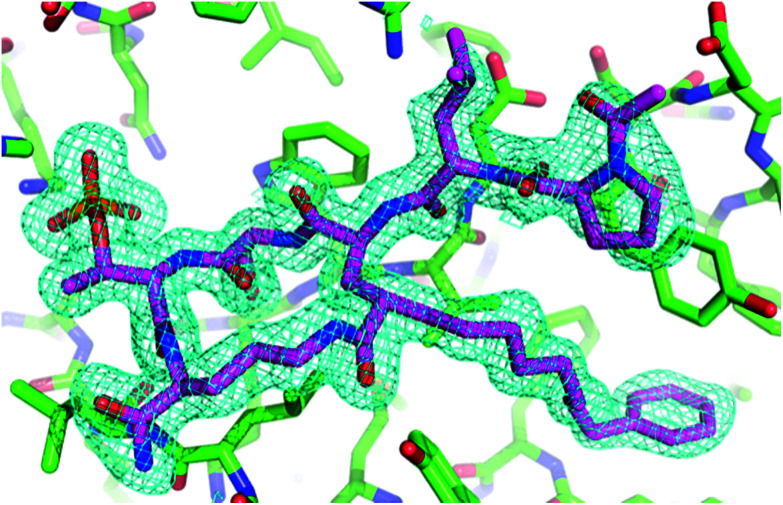
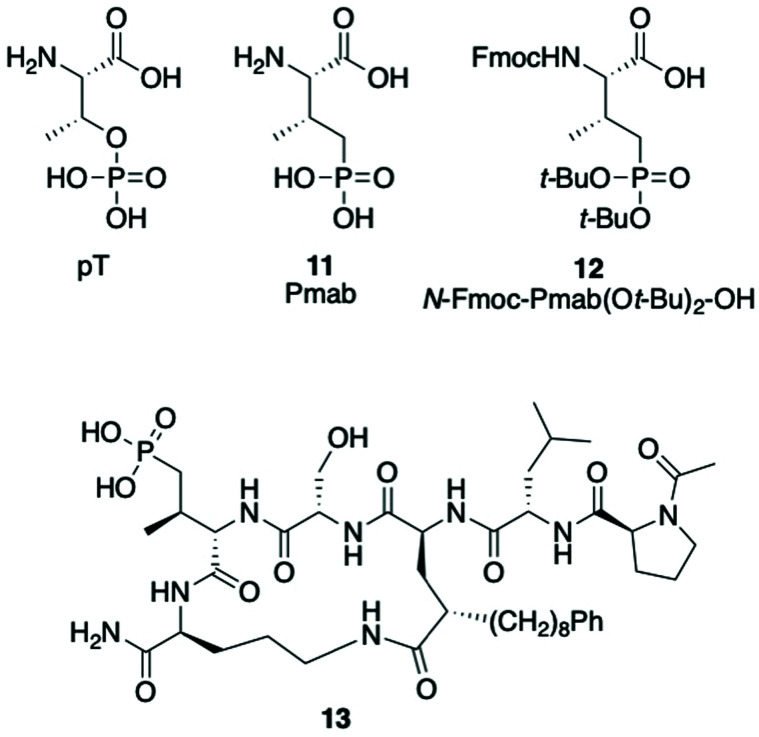
Targeting protein - protein interactions (PPIs) has emerged as an important area of discovery for anticancer therapeutic development. In the case of phospho-dependent PPIs, such as the polo-like kinase 1 (Plk1) polo-box domain (PBD), a phosphorylated protein residue can provide high-affinity recognition and binding to target protein hot spots. Developing antagonists of the Plk1 PBD can be particularly challenging if one relies solely on interactions within and proximal to the phospho-binding pocket. Fortunately, the affinity of phospho-dependent PPI antagonists can be significantly enhanced by taking advantage of interactions in both the phospho-binding site and hidden "cryptic" pockets that may be revealed on ligand binding. In our current paper, we describe the design and synthesis of macrocyclic peptide mimetics directed against the Plk1 PBD, which are characterized by a new glutamic acid analog that simultaneously serves as a ring-closing junction that provides accesses to a cryptic binding pocket, while at the same time achieving proper orientation of a phosphothreonine (pT) residue for optimal interaction in the signature phospho-binding pocket. Macrocycles prepared with this new amino acid analog introduce additional hydrogen-bonding interactions not found in the open-chain linear parent peptide. It is noteworthy that this new glutamic acid-based amino acid analog represents the first example of extremely high affinity ligands where access to the cryptic pocket from the pT-2 position is made possible with a residue that is not based on histidine. The concepts employed in the design and synthesis of these new macrocyclic peptide mimetics should be useful for further studies directed against the Plk1 PBD and potentially for ligands directed against other PPI targets.
靶向蛋白质-蛋白质相互作用(PPIs)已成为癌症治疗药物开发的一个重要发现领域。在磷酸依赖性 PPIs 的情况下,如 Polo 样激酶 1(Plk1) Polo 盒结构域(PBD),磷酸化蛋白质残基可以提供高亲和力的识别和结合靶蛋白热点。如果仅仅依赖于磷酸结合口袋内和附近的相互作用,开发 Plk1 PBD 的拮抗剂可能会特别具有挑战性。幸运的是,通过利用磷酸结合位点和隐藏的“隐匿”口袋中的相互作用,磷酸依赖性 PPI 拮抗剂的亲和力可以显著增强,这些口袋可能在配体结合时被揭示。在我们目前的论文中,我们描述了针对 Plk1 PBD 的大环肽模拟物的设计和合成,其特征是使用新的谷氨酸类似物,该类似物同时用作环封闭连接点,提供了进入隐匿结合口袋的通道,同时实现了磷酸苏氨酸(pT)残基的正确取向,以在特征性磷酸结合口袋中实现最佳相互作用。用这种新的氨基酸类似物制备的大环化合物引入了在开链线性母体肽中未发现的额外氢键相互作用。值得注意的是,这种新的基于谷氨酸的氨基酸类似物代表了第一个具有极高亲和力的配体的例子,其中通过不是基于组氨酸的残基从 pT-2 位置进入隐匿口袋成为可能。这些新的大环肽模拟物的设计和合成中采用的概念对于针对 Plk1 PBD 的进一步研究以及针对其他 PPI 靶标的配体应该是有用的。