Genomics Division, Instituto Tecnológico Y de Energías Renovables (ITER), Santa Cruz de Tenerife, Spain.
Estación Biológica de Doñana (EBD-CSIC), Integrative Ecology, 41092, Sevilla, Spain.
Sci Rep. 2021 Oct 15;11(1):20510. doi: 10.1038/s41598-021-99895-5.
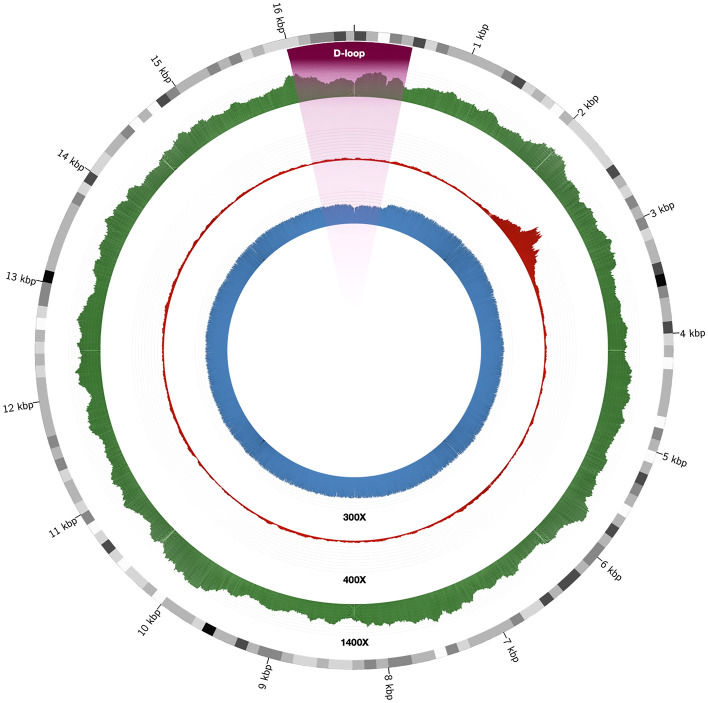
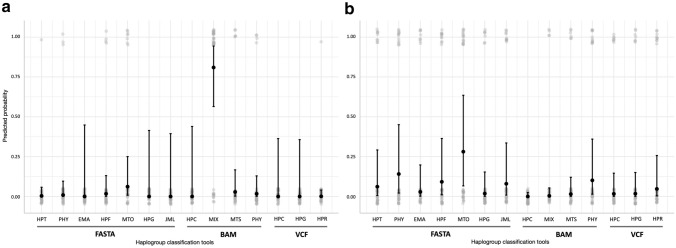
The mitochondrial genome (mtDNA) is of interest for a range of fields including evolutionary, forensic, and medical genetics. Human mitogenomes can be classified into evolutionary related haplogroups that provide ancestral information and pedigree relationships. Because of this and the advent of high-throughput sequencing (HTS) technology, there is a diversity of bioinformatic tools for haplogroup classification. We present a benchmarking of the 11 most salient tools for human mtDNA classification using empirical whole-genome (WGS) and whole-exome (WES) short-read sequencing data from 36 unrelated donors. We also assessed the best performing tool in third-generation long noisy read WGS data obtained with nanopore technology for a subset of the donors. We found that, for short-read WGS, most of the tools exhibit high accuracy for haplogroup classification irrespective of the input file used for the analysis. However, for short-read WES, Haplocheck and MixEmt were the most accurate tools. Based on the performance shown for WGS and WES, and the accompanying qualitative assessment, Haplocheck stands out as the most complete tool. For third-generation HTS data, we also showed that Haplocheck was able to accurately retrieve mtDNA haplogroups for all samples assessed, although only after following assembly-based approaches (either based on a referenced-based assembly or a hybrid de novo assembly). Taken together, our results provide guidance for researchers to select the most suitable tool to conduct the mtDNA analyses from HTS data.
线粒体基因组(mtDNA)在进化、法医和医学遗传学等多个领域都具有重要意义。人类线粒体基因组可以分为进化相关的单倍群,提供祖先信息和血统关系。由于这一点以及高通量测序(HTS)技术的出现,有各种各样的生物信息学工具可用于单倍群分类。我们使用来自 36 个无关供体的经验性全基因组(WGS)和全外显子组(WES)短读测序数据,对 11 种最显著的人类 mtDNA 分类工具进行了基准测试。我们还评估了最佳性能工具在第三代长噪声读取 WGS 数据中的表现,这些数据是使用纳米孔技术从一部分供体中获得的。我们发现,对于短读 WGS,大多数工具在进行单倍群分类时表现出很高的准确性,而不管用于分析的输入文件如何。然而,对于短读 WES,Haplocheck 和 MixEmt 是最准确的工具。基于 WGS 和 WES 的表现以及伴随的定性评估,Haplocheck 是最完整的工具。对于第三代 HTS 数据,我们还表明,Haplocheck 能够准确地检索所有评估样本的 mtDNA 单倍群,尽管这仅在遵循基于组装的方法(基于参考的组装或混合从头组装)之后才能实现。总之,我们的结果为研究人员提供了指导,以便从 HTS 数据中选择最适合进行 mtDNA 分析的工具。