Dipartimento di Chimica "Giacomo Ciamician", Alma Mater Studiorum-Università di Bologna, 40126 Bologna, Italy.
Int J Mol Sci. 2021 Oct 26;22(21):11567. doi: 10.3390/ijms222111567.
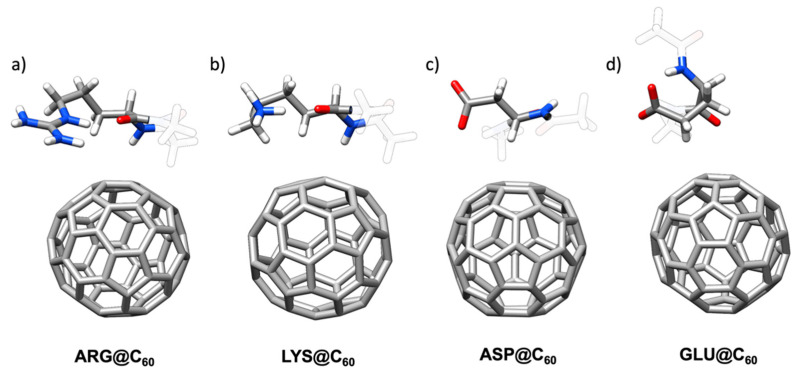
Molecular dynamics simulations were used to quantitatively investigate the interactions between the twenty proteinogenic amino acids and C. The conserved amino acid backbone gave a constant energetic interaction ~5.4 kcal mol, while the contribution to the binding due to the amino acid side chains was found to be up to ~5 kcal mol for tryptophan but lower, to a point where it was slightly destabilizing, for glutamic acid. The effects of the interplay between van der Waals, hydrophobic, and polar solvation interactions on the various aspects of the binding of the amino acids, which were grouped as aromatic, charged, polar and hydrophobic, are discussed. Although π-π interactions were dominant, surfactant-like and hydrophobic effects were also observed. In the molecular dynamics simulations, the interacting residues displayed a tendency to visit configurations (i.e., regions of the Ramachandran plot) that were absent when C was not present. The amino acid backbone assumed a "tepee-like" geometrical structure to maximize interactions with the fullerene cage. Well-defined conformations of the most interactive amino acids (Trp, Arg, Met) side chains were identified upon C binding.
采用分子动力学模拟方法定量研究了二十种蛋白氨基酸与 C 的相互作用。保守的氨基酸主链提供了约 5.4 kcal/mol 的恒定能量相互作用,而氨基酸侧链对结合的贡献则高达 5 kcal/mol,对于色氨酸而言,但对于谷氨酸而言,其贡献较低,甚至略微不利于稳定。讨论了范德华力、疏水性和极性溶剂化相互作用之间的相互作用对各种氨基酸结合的影响,这些氨基酸被分为芳香族、带电、极性和疏水性。尽管π-π相互作用占主导地位,但也观察到了表面活性剂样和疏水性效应。在分子动力学模拟中,相互作用的残基显示出倾向于访问配置(即 Ramachandran 图谱的区域)的趋势,而当 C 不存在时则不存在这些配置。氨基酸主链采用“锥形帐篷状”的几何结构,以最大化与富勒烯笼的相互作用。在 C 结合后,确定了最具相互作用的氨基酸(色氨酸、精氨酸、甲硫氨酸)侧链的明确构象。